
Au2Ag和AuAg2团簇对CO催化氧化反应的理论研究
2022-03-05 01:47陈金立
原子与分子物理学报 2022年3期
陈 宣 , 陈金立, 叶 影
(1.南京信息工程大学 物理与光电工程学院, 南京 210044; 2. 南京信息工程大学 电子与信息工程学院, 南京 210044)
1 前 言
近年来,一氧化碳氧化反应在开发一氧化碳传感器、提高二氧化碳激光器的效率、减缓氢燃料电池或生物燃料混合物的一氧化碳释放等方面的应用引起了人们极大的关注[1]. 自Haruta等人发现纳米金颗粒有着活跃的催化性质[2]以来,金团簇对CO催化氧化反应成为有一个十分活跃的研究领域[3-6]. CO催化氧化反应的关键时候O2分子的活性[7]. 金团簇可以很好的吸附CO分子,但对O2分子吸附和活化能力不是很强[8]. 许多实验和理论研究表明只有当金团簇中有未配对电子时,在吸附O2分子后,O2分子才能形成[O2]-结构[9, 10]. 因此金团簇的尺寸、离子态和氧化物载体将会有影响金团簇吸附O2分子的能力.
另一方面,一些金的合金团簇在一氧化碳氧化反应中展现的良好的催化性质[11-13]. 实验研究发现,Au-Ag纳米颗粒可以很好的催化CO氧化反应[14-16]. Guczi等人在实验中发现Au/Pd 团簇在TiO2载体上时可以有效的对CO进行催化氧化反应[17]. 在理论研究方面,55个原子的金银合金团簇和金铜合金团簇中, Au43Cu12团簇有很好的CO催化氧化性质[18],类似于小尺寸的Au10团簇,而Au25Ag30团簇的CO催化氧化能力也要比纯的Au55团簇和Ag55团簇高[19]. Peng 等人在理论研究AumPdn(m+n=3)团簇的CO 催化氧化反应中发现,Au2Pd 团簇的CO氧化反应势垒要低于Au3和Pd3团簇[20].
CO氧化反应有两种反应路径,分别为 Eley-Rideal(ER)反应机理和Langmuir-Hinshelwood(LH)反应机理[21-23]. 在ER反应机理中,CO气体直接与吸附在催化剂上的O2分子进行氧化反应. 而在HL反应机理中,CO和O2分子要共同吸附在催化剂上然后再发生氧化反应. 研究发现,W(111)表面、 W10、Cu6和Cu7团簇进行CO催化氧化反应时倾向于ER反应机理[24, 25],而Ni表面和Au55团簇则倾向于HL反应机理[26, 27]. 本文采用密度泛函理论研究了CO在Au2Ag和AuAg2团簇上的催化氧化反应. CO氧化反应中的两个反应过程,即CO+O2→ CO2+O和CO+O→CO2,都分别讨论了ER反应机理和HL反应机理. 我们将探讨CO氧化反应是否发生在团簇的Ag原子上?在Au2Ag和AuAg2团簇催化CO氧化反应中更倾向于那种反应机理?
2 计算方法
本文采用相对论密度泛函理论中的广义梯度近似(GGA)[28, 29],Perdew,Burke和Ernzerhof交换关联修正函数[30]和极化函数扩展的双数值原子轨道DND基组,也就是说函数中包含高于自由原子中的最高占据轨道角动量一级的角动量. 计算中采用自旋非限制近似求解Kohn-Sham[31]自洽场方程. 结构优化采用了Broyden-Fletcher-Goldfarb-Shanno方法,在没有任何参数限制(如对称性,键长,键角)条件下,以梯度变化小于10-3a.u、位移变化小于10-3a.u.和能量变化小于10-5a.u.作为收敛标准,自洽过程是在能量和电子密度的收敛标准为10-6a.u.下完成. 计算得出O2分子的键长为1.225 Å,CO分子的键长为1.143 Å,这与实验中测得的O2分子键长1.210 Å和CO分子键长1.130 Å[32]吻合的较好. 过渡态的搜索采用的是线性同步度越(linear synchronous transit, LST)和四极同步度越(quadratic synchronous transit, QST)方法.
3 结果与分析
3.1 Au2Ag和AuAg2团簇对O2和CO的吸附性质
图1给出了Au2Ag、AuAg2、Au2AgCO、Au2AgO2、AuAg2CO和AuAg2O2团簇的基态几何结构. Au2Ag和AuAg2的基态结构为平面等腰三角形. 我计算得到, Au2Ag团簇中Au-Au和Au-Ag的键长分别为2.834和2.679 Å, AuAg2团簇中Ag-Ag和Au-Ag 的键长分别为2.664和2.785 Å,这与之前的计算值吻合的较好[33]. 表1中列出了Au2AgCO、Au2AgO2、AuAg2CO和AuAg2O2中C-O和O-O的键长以及CO和O2吸附后的吸附能与CO和O2的Mulliken电荷分布. 吸附能为M团簇的能量与吸附物X的能量之和,减去吸附后MX团簇的总能量分(M= Au2Ag, AuAg2; X = CO, O2). 研究发现,O2分子倾向于吸附在有Ag原子的键位上. 在Au2Ag团簇中O2分子倾向于吸附在Au-Ag键上;在AuAg2团簇中,O2分子倾向于吸附在Ag-Ag键上. 而CO分子则倾向于吸附在Au原子的顶位上. 与自由O2分子键长1.225 Å相比,O2分子吸附后的O-O键长有了增长. Mulliken电荷分布指出O2分子在Au2AgO2和AuAg2O2中分别得到了0.354 e和0.414 e,这说明吸附后O2可能被活化. 相反的CO分子中的C-O键在吸附前后几乎没有变化. Mulliken电荷分布给出CO分子在Au2AgCO 和AuAg2CO中分别失去了0.213 e 和0.179 e. 此外,在AuAg2中,O2的吸附能(1.28 eV)比CO的吸附能(1.07 eV)大,表明在LH反应过程中应该是O2先吸附在Au2Ag团簇然后再进行CO + O2反应. 然而,在Au2Ag中,CO的吸附能(1.28 eV)比O2的吸附能(0.96 eV)大,说明在Au2Ag团簇上很难发生ER反应过程,但在氧气十分充足的情况下还是有可能发生反应的,并且在LH反应过程中应该是CO先吸附在Au2Ag团簇然后再进行CO + O2反应.
3.2 Au2Ag和AuAg2上CO + O2 → CO2 + O反应的ER和LH两种反应过程
基于上文中提到的吸附结果,图2和图3给出Au2Ag和AuAg2团簇上CO + O2→ CO2+ O反应的ER和LH两种反应过程. 表2列出了反应中各状态的结构参数. 在ER反应过程中,自由的CO分子靠近吸附在Au2Ag和AuAg2团簇上的O2分子,此时O-O键长分别为1.318 (IM1, 2b)和1.328 Å (IM1, 3b),然后直接反应,生成CO2并留下一个氧原子吸附在金属团簇上,反应能垒分别为0.86 (TS1-2, 2c) and 0.87 eV (TS1-2, 3c) .

表1 Au2AgCO、Au2AgO2、AuAg2CO和AuAg2O2中C-O和O-O键长,以及CO和O2吸附后的吸附能与CO和O2的Mulliken电荷分布.

图1 Au2Ag、AuAg2、Au2AgCO、Au2AgO2、AuAg2CO和AuAg2O2团簇的基态几何结构Fig. 1 The ground-state structures of Au2Ag, AuAg2, Au2AgCO, Au2AgO2, AuAg2CO, and AuAg2O2.
由上文可知,在LH反应过程中,Au2Ag团簇应先吸附CO分子,O2分子此时离Au2Ag较远,其距离为3.955 Å (IM1, 2f). 在IM1结构中,O2键长为1.240 Å, CO键长为1.154 Å. O2分子靠近Au2AgCO经过过度态TS1-2(2g),其反应势垒为0.04 eV. 此时O2到CO的距离(即O-C)为2.185 Å. 在IM2 (2h) 中,O2与CO之间形成一个弱键,O-C键长为1.607 Å,但是O2还没有与Au2Ag团簇形成键其之间距离为2.777 Å. 另外,O-O键增长到1.315 Å,但是C-O键长变化很小(1.185 Å). 在IM3结构中,O-O键继续变长并增长到1.516 Å, O-Au和O-C键长则分别缩短到2.168和1.318 Å,C-O键长仍然变化很小. 从IM2到IM3,反应势垒为0.28 eV,其相应的过渡态为TS2-3(2i). 显然,经过IM3状态后,如果O-O键继续拉长断开,然后就会形成新的OCO (CO2). TS3-4(2k)结构中,O-O键长增长到1.743 Å,O-C键长缩短为1.266 Å,这长度接近与气相CO2的O-C键长1.176 Å ,其反应势垒为0.33 eV. 经过TS3-4,新的CO2分子形成并远离Au原子,留下一个O原子在Au原子上.
AuAg2的LH反应过程应为O2先吸附在AuAg2团簇上,然后CO靠近AuAg2O2团簇并吸附在Au原子上,形成了共吸附状态O2- AuAg2-CO (IM1, 3f). 在IM1结构中,O2吸附在Ag-Ag键上,其键长为1.319 Å,而CO吸附在Au原子上,其键长为1.151 Å. 如果要发生氧化反应,O2和CO需要彼此靠近. O2从Ag-Ag键移动到一个Ag原子上时,形成了一个稳定的结构 (IM2, 3h).在O2移动的过程中,其反应势垒为0.29 eV,相应的过渡态为TS1-2(3g). 另外IM1, IM2和TS1-2结构中,O-O键和C-O键长几乎没有变化. 在O2和CO相互靠近的过程中经过了势垒为0.40 eV 的过渡态TS2-3(3i),此时O2和CO之间的距离(O-C)为2.537 Å. 在IM3 (3j)结构中,O-C键长缩短为2.008 Å,但O-O和C-O键长与 TS2-3结构中的相应键长相比变化不大. O2和CO继续靠近彼此,经过过度态TS3-4(3k),其势垒为0.16 eV. 在TS3-4结构中,O-C键长大幅度的缩短为1.582 Å,并且O-O和C-O键长有少许增长. 在IM4 (3l)结构中,AuAg2团簇上形成了稳定的OOCO吸附物,其O-C键长缩短到1.355 Å,而O-O和C-O键长分别增长到1.434和1.210 Å. 从IM4经过了势垒为0.68 eV的过渡态TS4-5(3m),其O-O键断开, O-C键正在形成,最终形成 IM5 (3n).
总体上看,在Au2Ag和AuAg2团簇上CO + O2→ CO2+ O的LH反应过程中,OCOO 结构中O-O键的断裂是反应的关键,相应的反应势垒分别为0.33 (TS3-4, 2k) 和0.68 eV (TS4-5, 3m). 另外在CO + O2→ CO2+ O反应中,反应能垒明显指出LH过程比ER过程优越,这可能是因为OCOO 结构中O-O键活化度比较高. 在LH过程中,Au2Ag 和AuAg2团簇上会形成OCOO 结构,相比于自由O2分子键长,OCOO 结构中的O-O键长分别被拉长了23.7% (IM3, 2j)和17.1% (IM4, 3l) ,这将促使O-O键的断裂和CO2分子的形成. 然而,在ER过程中,O2吸附在Au2Ag 和AuAg2团簇上后,O-O键是有所增长,但于自由O2分子键长相比,仅仅增长了7.4% (IM1, 2b) 和8.4% (IM1, 3b) ,这就导致ER反应的反应能垒较高.
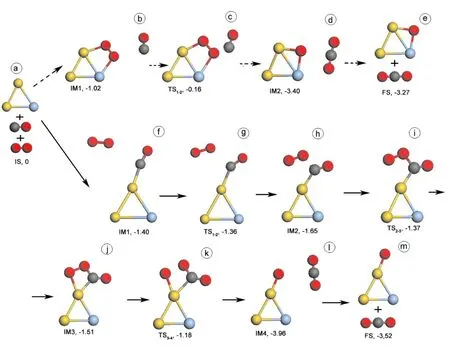
图2 在Au2Ag团簇上CO + O2→ CO2 + O两种反应过程中各状态对应的几何结构和相应能量,虚箭头为ER反应过程,实箭头为LH反应过程,其中金属为Au原子,蓝色为Ag原子,红色为O原子,灰色为C原子.Fig. 2 Structures of initial state (IS), intermediates (IM), transition states (TS), final states (FS), and relative energy (eV) with respect to IS in the paths which CO is oxidized by molecular O2 on Au2Ag via ER mechanism (dash arrow) and LH mechanism (solid arrow). The yellow, blue, gray, and red balls denote Au, Ag, C, and O atoms, respectively.

表2 在Au2Ag和AuAg2团簇上CO + O2→ CO2 + O两种反应过程中不同状态的结构参数,其中O-O为O2分子中两个氧原子之间的距离,O-C为O2与CO之间的距离,C-O为CO分子中C原子与O原子之间的距离.
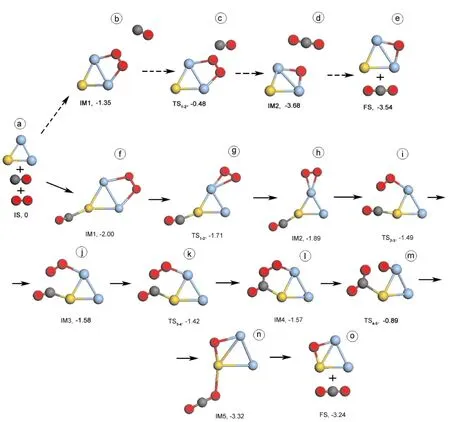
图3 在AuAg2团簇上CO + O2→ CO2 + O两种反应过程中各状态对应的几何结构和相应能量,虚箭头为ER反应过程,实箭头为LH反应过程,其中金属为Au原子,蓝色为Ag原子,红色为O原子,灰色为C原子.Fig. 3 Structures of initial state (IS), intermediates (IM), transition states (TS), final states (FS), and relative energy (eV) with respect to IS in the paths which CO is oxidized by molecular O2 on AuAg2 via ER mechanism (dash arrow) and LH mechanism (solid arrow). The yellow, blue, gray, and red balls denote Au, Ag, C, and O atoms, respectively.
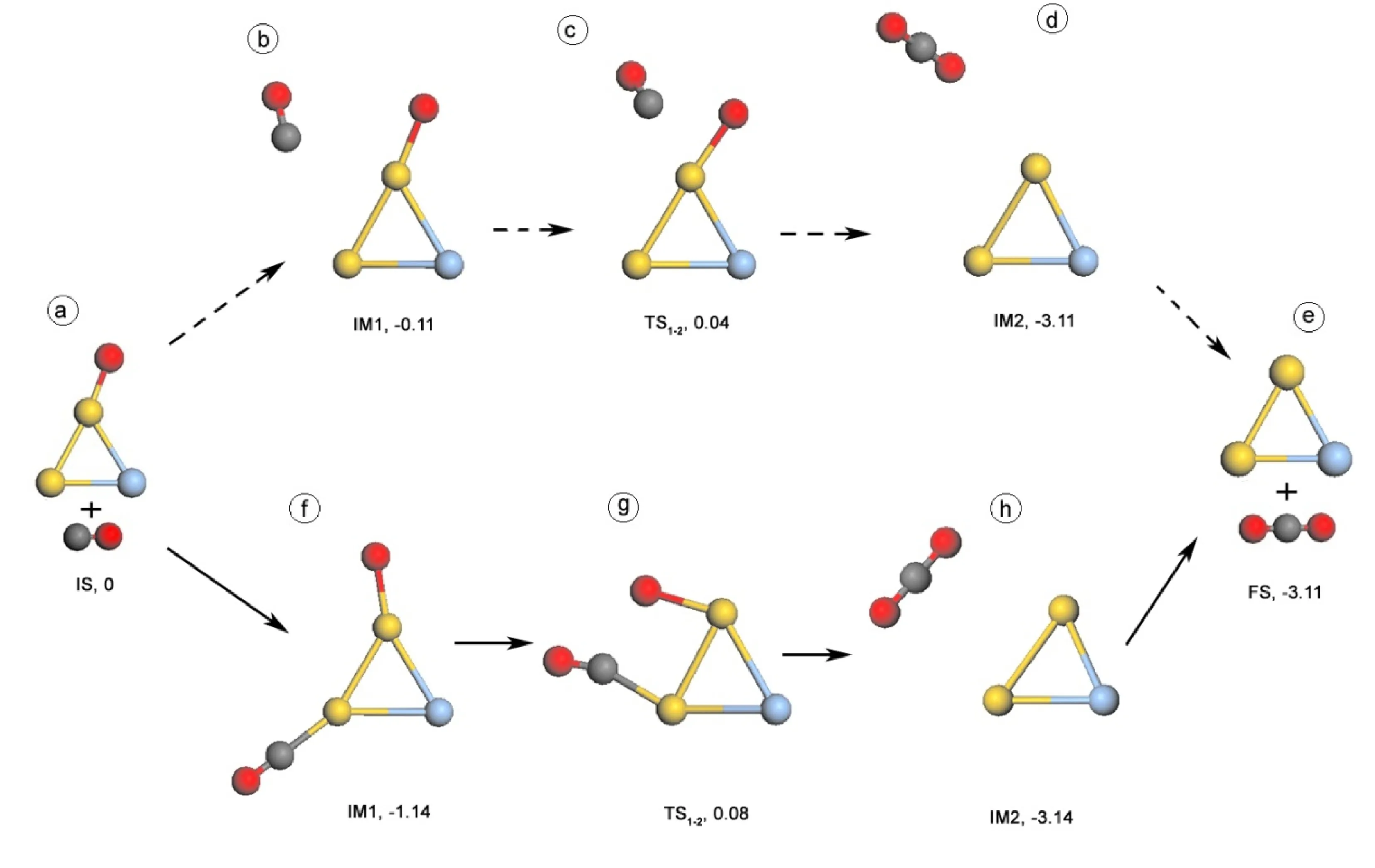
图4 在Au2Ag团簇上CO + O → CO2两种反应过程中各状态对应的几何结构和相应能量,虚箭头为ER反应过程,实箭头为LH反应过程,其中金属为Au原子,蓝色为Ag原子,红色为O原子,灰色为C原子.Fig. 4 Structures of initial state (IS), intermediates (IM), transition states (TS), final states (FS), and relative energy (eV) with respect to IS in the paths which CO is oxidized by O atom on Au2Ag via ER mechanism (dash arrow) and LH mechanism (solid arrow). The yellow, blue, gray, and red balls denote Au, Ag, C, and O atoms, respectively.
3.3 Au2Ag和AuAg2上CO + O → CO2的ER和LH两种反应过程
CO + O→ CO2在Au2Ag和AuAg2团簇上的反应我们也考虑了ER和LH两种反应过程. 图4和图5给出Au2Ag和AuAg2团簇上CO + O → CO2反应的ER和LH两种反应过程. 表3列出了反应中各状态的结构参数. 由上文可知,CO + O2→ CO2+ O反应中,LH过程优于ER过程. 因此,CO + O → CO2在Au2Ag和AuAg2团簇上的反应的初始结构为CO + O2→ CO2+ O反应的LH过程的最终结构,即Au2Ag中O原子吸附在一个Au原子上,AuAg2中O原子吸附在Au-Ag键上.
在ER反应过程中,CO从远处靠近Au2AgO和AuAg2O团簇,然后直接与O原子进行反应生成CO2,其能垒分别为0.15 (TS1-2, 4c)和0.02 eV(TS1-2, 5c). 在LH反应过程中,CO分子和O原子之间在Au2AgO和AuAg2O团簇上相互靠近是反应的关键,其势垒分别为1.06 (TS1-2, 4g)和1.63 eV (TS1-2, 5g) . 因此,CO + O → CO2在Au2Ag和AuAg2团簇上的反应过程中,ER过程优于LH过程. 这可能是因为LH过程中CO与O之间相互靠近要经过较远的距离和较大的角度改变. 比如,O和CO共吸附在Au2Ag团簇的两个不同的Au原子上,O-C之间的距离为5.965 Å (IM1, 4f);而O和CO共吸附在AuAg2团簇上时,虽然CO和O都吸附在相同的Au原子上,但O-C距离为3.829 Å (IM, 5f),并且吸附的位置正好相反∠CAuO为174°,由于位阻效应使得CO很难靠近O原子,从而导致较大的反应能垒.

图5 在AuAg2团簇上CO + O → CO2两种反应过程中各状态对应的几何结构和相应能量,虚箭头为ER反应过程,实箭头为LH反应过程,其中金属为Au原子,蓝色为Ag原子,红色为O原子,灰色为C原子.Fig. 5 Structures of initial state (IS), intermediates (IM), transition states (TS), final states (FS), and relative energy (eV) with respect to IS in the paths which CO is oxidized by O atom on AuAg2 via ER mechanism (dash arrow) and LH mechanism (solid arrow). The yellow, blue, gray, and red balls denote Au, Ag, C, and O atoms, respectively.
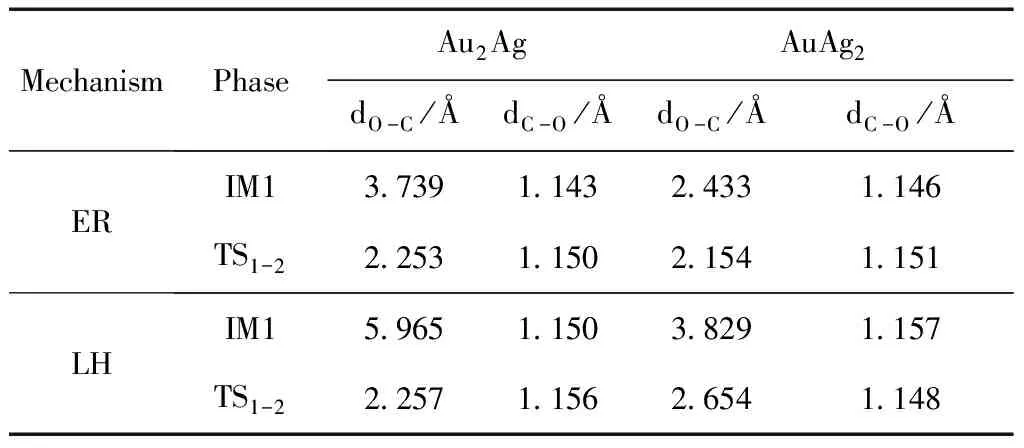
表3 在Au2Ag和AuAg2团簇上CO + O → CO2两种反应过程中不同状态的结构参数(Å),其中O-C为O与CO之间的距离,C-O为CO分子中C原子与O原子之间的距离.
4 结 论
本文采用密度泛函理论研究了CO在Au2Ag和AuAg2团簇上的催化氧化反应机理. 研究发现:在CO + O2→ CO2+ O反应中,虽然O2分子吸附在Au2Ag和AuAg2团簇上后被活化,但被活化的程度不够,导致ER反应的反应能垒较高,反应过程更倾向于LH反应. 而在CO + O → CO2反应中,由于位阻相应导致LH反应的反应能垒较高,反应过程更倾向于ER反应. 所以,完整的CO在Au2Ag和AuAg2团簇上的催化氧化反应为先经过LH反应过程,再经过ER反应过程. 由反应势能可知,CO在Au2Ag团簇上的两个反应过程的反应势能都较低,因此Au2Ag团簇有望成为良好的CO氧化催化剂.
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
建材发展导向(2021年14期)2021-08-23
当代陕西(2020年23期)2021-01-07
智富时代(2018年8期)2018-09-28
智富时代(2018年8期)2018-09-28
冰雪运动(2016年6期)2016-05-17
小朋友·快乐手工(2016年9期)2016-05-14
数学大世界·小学低年级辅导版(2009年8期)2009-07-28
