
Ab initio simulations of NO adsorption on hematite(0001)surface: PBE versus PBE+U*
2022-03-19 01:37WUCuixiaSUNTaoFABRISStefanoDULin
中国科学院大学学报 2022年2期
WU Cuixia,SUN Tao†,FABRIS Stefano,DU Lin
(1 College of Earth and Planetary Sciences, University of Chinese Academy of Sciences,Beijing 100049, China;2 CNR-IOM DEMOCRITOS, Istituto Officina dei Materiali, Consiglio Nazionale delle Ricerche, Trieste 34136, Italy;3 Environment Research Institute, Shandong University, Qingdao 266237, Shandong, China)(Received 24 July 2020;Revised 10 August 2020)
Abstract NOx(x=1,2)are major air-pollutants detrimental to human health and much effort has been devoted to find efficient photocatalysts capable of removing NOx from air(de-NOx).Recent experiments indicate that hematite(α-Fe2O3)is a promising de-NOx photocatalyst.However some key features of the NO adsorption on the hematite surface remain unclear, hindering further comprehension of the photocatalytic process.Here we study the adsorption of NO on the hematite(0001)surface using the PBE+U method with a dispersion correction(vdw)in the framework of density functional theory(DFT).We find the addition of a Hubbard U term in the DFT Hamiltonian strongly affects the adsorption properties, with the adsorption energy(-0.64 eV)decreased by 50% with respect to those of PBE(-1.31 eV).This decrease is attributed to two factors:(i)the U term shifts the energy of Fe 3d orbitals away from the valence band maximum, making them chemically less active;(ii)the NO molecule has an unpaired π* electron and is more sensitive to the electronic structure of the substrate.In contrast to the inclusion of U, the dispersion correction causes little change to the adsorption properties except increases the adsorption energy by about-0.18 eV.We use the Langmuir formula to calculate the thermal equilibrium coverage of NO on the hematite(0001)surface and find predictions made with the PBE+U vdw are more consistent with experiments.These results highlight the importance of strong electronic correlations in describing the hematite surface reactions, and may serve as a starting point to unravel the complete photocatalytic mechanism.
Keywords hematite; NO; photocatalysis; Langmuir; air pollution
NOx(x=1, 2)are major air pollutants in urban areas originated from fossil fuel combustions in vehicles and power stations[1-2].They are detrimental to human health and much effort has been devoted to remove them from air(de-NOx).Among the various de-NOxtechnologies under development, photocatalytic oxidation(PCO)is one of the most promising routines[3-4]: under sunlight, a photocatalytic substrate captures NOxin the air and transforms them into non-volatile nitrates.These nitrates can then be rinsed away and the substrate regains its photocatalytic ability.So far the most common photocatalytic de-NOxmaterial is TiO2, with TiO2-based air purification devices being deployed in several European cities[4].However due to its relatively large band-gap, TiO2can only absorb ultra-violet light, or about 4%-5% of the total sunlight energy[5-6], therefore there is a strong initiative to find more efficient catalysts.Hematite(α-Fe2O3)is considered a good alternative[7-9]to TiO2because of its narrower band-gap, high stability, non-toxicity, and low cost.Indeed, hematite-based nanostructures has recently been found[9]to exhibit outstanding de-NOxabilities, with both the NO conversion efficiency and selectivity comparable or superior than commercial TiO2catalysts.To further improve the catalytic efficiency of hematite, detailed knowledge on its PCO mechanism is essential.The adsorption of NO molecules on the hematite surface is a key step in the PCO process, however some of its main features remain unclear, calling for further investigations.
Computer simulations based on density functional theory(DFT)is a powerful method and in principle well-suited to determine the atomistic adsorption properties[10-15], yet for the NO-hematite system existing reports are conflicting and inconclusive.Song et al.[16]conducted DFT simulations using the Perdew-Burke-Ernzerhof(PBE)exchange-correlation functional[17].They found that stable chemisorption took place only at the surface Fe sites(denoted as Fe*)with an adsorption energy of-1.31 eV.In contrast, Li et al.[18]included dispersion corrections(PBE vdw)and determined the adsorption energy as-4.08 eV.Such a large difference is surprising and its origin remains unclear.Moreover, both adsorption energies are quite high, indicating that NO can easily adsorb on the α-Fe2O3surface.This seems in conflict with experimental observations[7,9]where no infrared signals from adsorbed NO molecules were detected when the illumination was off and the NO concentration was low(1×10-7).Note α-Fe2O3is a typical transition metal oxide where the d-electrons of iron atoms exhibit strong correlations.For such systems the PBE or PBE vdw methods may not be suitable and more advanced techniques such as the PBE+U[19]or hybrid functionals[20]are needed.
Here we use the PBE+U method with dispersion corrections(vdw)to study NO adsorption on the α-Fe2O3(0001)surface.The effects of Hubbard U and vdw were carefully delineated.The microscopic bonding mechanism was determined by analyzing the partial density of states(pdos)and charge density differences before and after NO adsorption.Finally, we applied the Langmuir formula from statistical mechanics to predict the equilibrium coverage as a function of NO concentration in the air.
1 Computational details
Calculations were performed using the plane-wave pseudopotential method as implemented in the open-source QUANTUM-ESPRESSO package[21].To avoid uncertainties associated with semi-core electrons, we use an ultrasoft pseudopotential[22]with 16 valence electrons(3s23p63d64s2)to describe Fe, N and O were described using ultrasoft pseudopotentials with 2s22p3and 2s22p4valence electrons, respectively.Previous studies have long established that physical properties of hematite are best described by the PBE+U method withU=4.2 eV[23-27], thus we adopt the sameUvalue here.NO is a polar molecule and dipole-dipole interactions may substantially affect its adsorption properties.However such dipole interactions were absent in the standard PBE functional, thus a semi-empirical dispersion correction(vdw)[28]was added to the simulation.The energy cutoff of the plane-wave basis was set to 40 Ry, and the Brillouin zone sampling was conducted at 4×4×2 for the hematite bulk, and 4×4×1 for the slab.With these settings the adsorption energies were converged within 0.01 eV.To ensure the reliability of the calculations, a tight energy convergence criterion(10-8Ry)was set for the electron self-consistent field(scf)calculations.Ionic relaxations were performed until every force component on the atoms was less than 0.2 mRy/bohr(50 meV/nm).
α-Fe2O3maintains a R-3c, corundum structure and is anti-ferromagnetic along the c-axis.Figure 1 shows the slab supercell we are using, where yellow and white arrows distinguish the spin-up and spin-down iron atoms.The surface corresponds to the Fe-O3-Fe termination, which has been identified as thermodynamically most stable[19,24]at ambient conditions.The slabs were separated by a 24 Å(1 Å=0.1 nm)vacuum layer so as to minimize their interactions.To delineate the effect of +U and vdw, structural relaxations were performed with four different settings: PBE, PBE vdw, PBE+U, and PBE+U vdw.During the structural relaxation, the atoms in the middle three layers were fixed at their bulk positions, whereas all other atoms were allowed to move.
Once structural relaxations were completed, we evaluated the adsorption energyEadas follows
Ead=Em+s-(Em+Es),
(1)
whereEm+s,Em,andEsare the energies of the adsorbed system, isolated molecule, and the slab, respectively.A negativeEadindicates the adsorption is exothermic: the greater the absolute value ofEad,the more stable the adsorbed configuration.
To understand the effect of coverage on the adsorption, we first performed the simulation in the 1×1 supercell(100% monolayer coverage), then repeated the simulation in the 2×2 supercell(25% monolayer coverage).The resulting adsorption geometries and energies are nearly identical.In fact, as the distance between adjacent surface Fe*atoms are more than 5 Å, the interactions among adsorbates are small even at full coverage.
2 Results and discussion
2.1 Adsorption geometries and energies
Prior studies have identified that the most stable NO adsorption configuration as the N atom pointing downwards near vertically to a surface Fe*atom[16,18], with the N-Fe*bond length about 1.7 Å.We took this configuration as the initial coordinates and performed structure relaxations under four different settings: PBE, PBE vdw, PBE+U and PBE+U vdw.The resulting adsorption geometries are shown in Fig.2.Comparisons with prior studies are listed in Table 1.Our PBE results agree very well with those of Song et al.[16], whereas our PBE vdw results differ substantially with Li et al.[18]The adsorption energies of PBE and PBE vdw from our calculations differ by~0.2 eV, in accordance with the typical range of van der Waals energies[28].By contrast, the contribution the van der Waals interaction would be 2.77 eV if the adsorption energy of PBE vdw is indeed-4.08 eV[18].Such a large van der Waals contribution is unreasonable as it exceeds the bond strengths of many covalent bonds.The PBE vdw results obtained from this study is consistent with those of PBE and should be more reliable.

Fig.2 Adsorption geometries of NO on hematite(0001) surface predicted with four different settings
More importantly, we find that the addition of a Hubbard U term in the DFT Hamiltonic affects strongly the adsorption properties of NO on hematite.Once the U is turned on, the N-Fe*bond length gets elongated by as much as 0.3 Å.The inclination of the NO molecule also changes substantially, from nearly vertical to the surface(∠O-N-Fe*=170°)to an O-N-Fe*angle of about 125°.The adsorption energy decreases by~50%, from-1.31 eV to-0.64 eV.This trend is unaffected by the inclusion of dispersion correction(vdw), whose overall effect is to increase the adsorption energy by~0.18 eV while causes little changes in the adsorption geometries.It has been known that for transition metal oxides[29]the DFT+U can predict adsorption properties different from those of standard DFT.However to our knowledge this is yet to be reported for NO-hematite.In fact, in our previous study on the SO2adsorption on the hematite surface[27], the adsorption energy was found to be nearly independent ofU, thus it is surprising to see NO behaves so differently.
2.2 Electronic structures

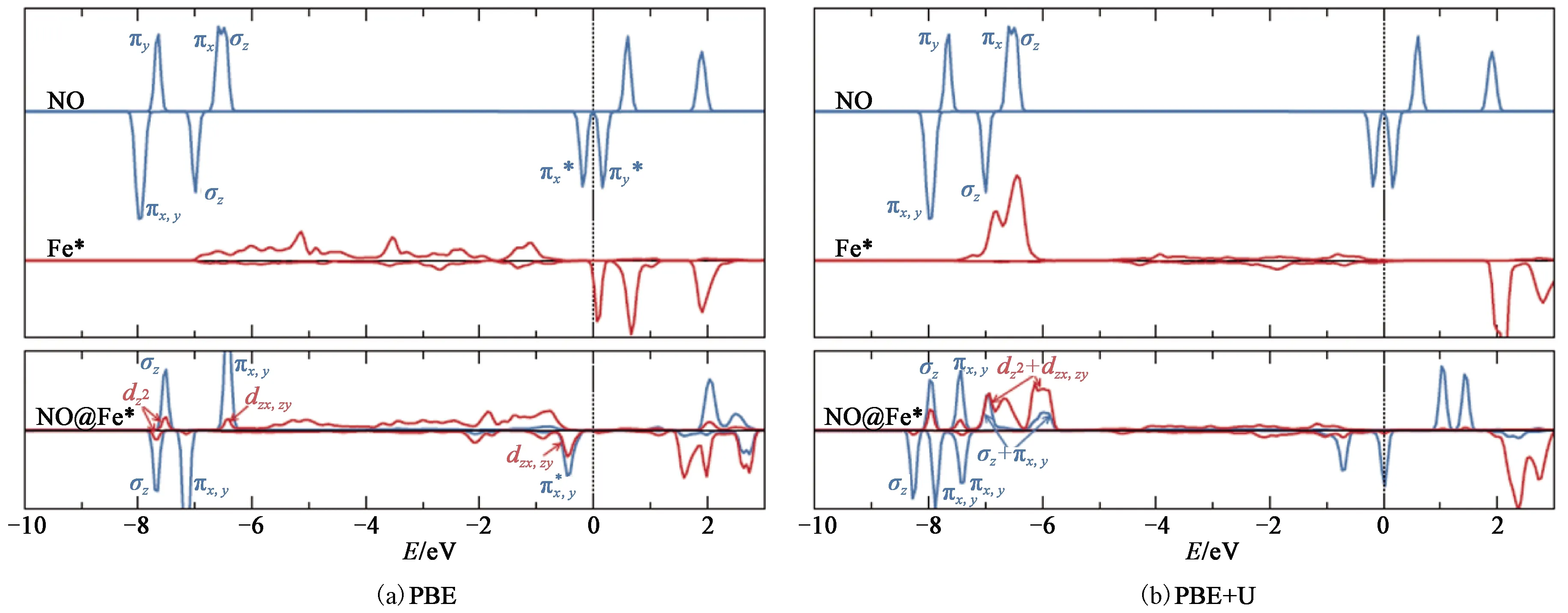
Fig.3 Partial density of states of the NO molecule and the surface Fe atom(Fe*)before(upper panel) and after(lower panel)adsorption using(a)PBE and(b)PBE+U
Further insights can be obtained by examining the charge density difference with and without Hubbard-like U correction, as shown in Fig.4.Upon adsorption, significant charge accumulations take place in the region between the N and the Fe*atoms, indicating that the adsorption is mainly driven by forming new covalent bonds rather than charge transfer reactions such as Fe3++ NO = NO++ Fe2+.Indeed, the integrated charge difference in the region occupied by the NO molecule is less than 0.01e.By contrast, the integrated charge difference in the region between the N and the Fe*atoms is 0.18 e(PBE)and 0.08 e(PBE+U), respectively.The less covalent charge predicted in the PBE+U calculation is consistent with its smaller adsorption energy.

The iso-surface value is ±0.02 e/Å3.
We now consider the effect of dispersion correction(vdw)on the electronic structures.This semi-empirical correction is not involved in the scf cycles to determine the electronic ground state, yet it can exert an indirect influence on the electronic structure by changing the final relaxed coordinates.As the changes in the relaxed coordinates are small(see Table 1), we find the associated changes in the electronic structure are minor.Still, as dipole-dipole interactions are present in real systems, we use the adsorption properties evaluated with vdw to compare with experiments.
2.3 Equilibrium coverage
We now apply the information obtained in prior sections to calculate the equilibrium coverage of NO on hematite.Assume a mono-layer adsorption where the adsorption configuration at each adsorption site is identical and the adsorbates do not interact, the thermal equilibrium coverage of the adsorbed molecule can be expressed in terms of the Langmuir formula[32]as
(2)

(3)
where ΔGm(T,P0)is the Gibbs energy change of the gas molecule from 0 K to T at the standard atmospheric pressure.For NO it equals-0.56 eV at 298.15 K[33].The resulting Θ(P)are shown in Fig.5.As theEadfrom PBE vdw is high(-1.51 eV), substantial amount of NO will be adsorbed on the surface even when the NO partial pressure is as low as 10-12Pa.In fact, a full coverage is achieved whenP>10-9Pa.By contrast, theEadfrom PBE+U vdw is low(-0.82 eV)and a full coverage can be achieved only whenP>100 Pa.In the experiments[7,9], no infrared signal from the adsorbed NO molecules were observed when the light was off and the NO concentration is 1×10-7(P=0.01 Pa), indicating that the amount of adsorbates is scarce under such conditions.By contrast, distinct infrared signals from the adsorbed NO molecules were detected when the NO partial pressures were greater than 5 Torr(667 Pa)[34].These observations are consistent with the Θ(P)predicted by PBE+U vdw while in conflict with that of PBE vdw.We therefore conclude that including the Hubbard U provides a more accurate description of the system.

Fig.5 Equilibrium Coverage of NO molecule on the Hematite(0001)surface at 298.15 K
The above analysis considers only one type of gas molecule(NO)adsorbing on the hematite surface.This is appropriate when the illumination is off.When the illumination is on, electron-hole pairs will be generated at the hematite surface.These electron-hole pairs will interact with the O2, H2O molecules in the air and convert NO into thermodynamically more stable compounds such as NO3.The formation of NO3is crucial in trapping significant amount of NO molecules on the hematite surface, otherwise for typical NO concentrations in the atmosphere, the number of adsorbed NO molecules would be scarce.The exact mechanism of this de-NOxprocess, as well as how to use this mechanism to further improve the catalytic efficiency of hematite, will be a focus for future research.
3 Conclusions
We have studied the adsorption of NO molecules on the hematite(0001)surface using DFT.We found that the inclusion of a Hubbard term in the DFT Hamiltonian strongly affects the NO adsorption geometries and energies.The smaller adsorption energies predicted with +U yield equilibrium coverages that are more consistent with experimental observations.As this U-dependence is related to the unpaired electron of the NO molecule, we anticipate similar behavior in other molecules such as NO2, which also possess lone electrons.In contrast, the effect of dispersion correction is not as dramatic as previous studies may indicate, as the adsorption geometries and electronic structures with or without the vdw correction are nearly identical.This work clarifies the mechanism of the NO adsorption, and paves the way for further work to fully understand the de-NOxprocess on the hematite surface.
