
基于QCL吸收光谱的SO2,SO3同步测量技术
2022-06-09 05:03俞峰苹李济东彭志敏尤晨昱赵金龙
激光与红外 2022年5期
俞峰苹,李济东,彭志敏,尤晨昱,赵金龙
(1.浙江天地环保科技股份有限公司,浙江 杭州 311100;2.清华大学能源与动力工程系,北京 100084)
1 引 言
在含硫燃料的燃烧过程和废气排放中,SO2和SO3是主要的污染物,是空气污染治理中重要的一环。SO2生成于多种过程中,例如化石燃料的燃烧和硫酸盐的热分解。SO3在自然界中无法稳定存在,极易溶于水生成H2SO4,因此也被称作硫酸或硫酸雾。SO3可由SO2在高温条件下的均质气相反应或非均质催化反应产生。由于SO2和SO3具有较强的酸性和腐蚀性,因此有害于人类的身体健康,尤其是人类的呼吸系统和眼睛[1]。在工业现场,SO3还易提高烟气露点温度造成低温腐蚀,从而导致工厂设备损坏。另外,SO3是酸雨的主要成分,会毁坏森林和水生生物,并造成城市建材和涂料的腐蚀[2]。因此,在含硫混合气体中对SO2和SO3的监测尤为重要。
目前用于工业环境监测的SO2测量方法有很多,其中光学测量技术具有响应快、灵敏度高和可实时在线测量的优势。常见的光学测量技术包括非分散红外吸收光谱技术、紫外差分吸收光谱技术、非分散紫外吸收光谱技术、高温红外吸收光谱技术、紫外荧光技术和干涉分光技术等[3-14]。
SO3由于在500 ℃以下时极易溶于水生成硫酸,且室温下为液态,标况下为固态,因此其精确测量比SO2更具有挑战性,其测量通常基于SO42-的间接测量,首先进行烟气采样,将样气中的SO3转化为SO42-,再通过离子色谱法、分光光度计法、比浊法、滴定法等检测手段测定SO42-含量,从而间接得出SO3浓度。采样方法包括控制冷凝法、异丙醇吸收法、酸露点测量法和硫酸盐置换法等[15-19],但是间接测量法很难实现实时在线监测且取样过程中SO3的损失也加大了测量误差。另外,在这些间接测量方法尤其在异丙醇吸收法中,高浓度SO2的干扰成为关键问题。基于吸收光谱的光学传感器也已经应用于SO3测量,例如傅里叶红外光谱(FITR)和紫外吸收光谱[20]。由于这些光学测量方法需要提前在SO3浓度已知标准气体中标定SO3的吸收特性,但SO3标气制备难度较大[21-23],因此这些方法很难实现SO3的定量测量。更重要的是,SO3的吸收光谱结构即使在低压条件下也呈现连续谱,且与SO2光谱严重耦合,因此在有限的光谱分辨率下很难在混合气体中得到SO3的特征吸收光谱。
激光吸收光谱技术目前已应用于气体物性参数测量[24-26],例如在文献[27]中,美国Wilson教授等人采用两支7~8 μm的QCL激光器在燃烧室尾气中同时测量SO2和SO3。芬兰Hieta等人采用QCL激光器结合FTIR同时测量SO2,SO3和H2O三种组分[28]。然而,由于设备振动或污染引起的基线(背景信号)漂移限制了测量SO3吸收率的准确性。最重要的是,SO3高温光谱数据库的缺乏使得研究者们无法从混合气体光谱中通过同步拟合修正基线并得到准确的吸收率。SO3在4 μm波段的吸收谱线也被应用于SO3浓度测量,在文献[29]中,日本Tokura教授采用基于差频合成的中红外激光技术借助4 μm谱线测量SO3浓度。尽管H2O,CO2等燃烧产物的吸收光谱在4 μm波段处对SO3光谱干扰较小,但该波长对应的ν1+ν3振动带吸收强度远弱于7 μm波长对应的ν4振动带,因此检测下限较低。
本文描述了基于7.16 μm QCL吸收光谱的高温条件下SO2和SO3同步测量系统。SO3气体由SO2催化氧化制取并在反应过程中完成两种气体浓度的测量,从而计算得到不同温度和压力条件下的反应转化率。本文通过修正SO2和SO3的高温光谱模型,成功将两者的独立光谱结构从严重耦合的混合气体光谱中解耦并用于测量气体浓度。同时,本文还探究了SO2在钒基催化剂表面的吸附效应随压力的变化关系。另外,在该实验系统中还通过设计冷却吸收装置将反应后的SO2和SO3气体分离并测量剩余SO2浓度,间接计算得到反应转化率,从而进一步验证反应过程中同步测量的准确性。
2 吸收光谱理论
单色光在频率ν处的透过率,τ(ν),在通过均匀吸收气体介质后的表达式为:
(1)
其中,I0和It分别是入射和出射激光强度;α(ν)是光谱吸收率,对于独立谱线的邻近区域内,α(ν)与气体的热物性参数相关,其表达式如下:
α(ν)=PS(T)φ(ν)χL
(2)
其中,S(T)[cm-2atm-1]是谱线线强;P[atm]是气体压力;χ是待测气体的摩尔分数;L[cm]是测量光程;φ(ν)[cm-1]是线型函数(线型模型)。
SO2和 SO3的独立谱线线型,φ(ν),在本文中采用Voigt模型描述。Voigt模型是Lorentz和Gauss 模型的卷积模式,同时将碰撞展宽和多普勒展宽考虑在内,其函数表达式如下[30-31]:
(3)
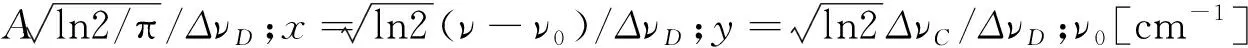
(4)
其中,M[g·mol-1]是待测气体分子的相对分子质量。谱线碰撞展宽与分子间碰撞频率正相关,其函数表达式如下:
(5)
其中,γA-B(T)[cm-1atm-1]是温度为T的条件下背景气体A和待测气体B之间的碰撞展宽系数;χB是待测气体的摩尔分数。
3 实验装置和步骤
图1所示的SOx测量实验装置主体由高温管式炉内的两腔(空腔、催化腔)一室(光学高温测量气室)组成。上游连接气体流动控制系统和气体混合系统,下游连接真空泵用于驱动和维持实验过程中的气体流动。位于高温管式炉(Kejia Furnace,KJ-200827T)中的机械结构主要由三部分组成,处于中心位置的是高温测量气室,其表面固定有3支精度为0.75 %的热电偶探头(Omega,TJ80-CAXL-116U),用于测量3个位置处的实验温度。其两侧装配有带有2°楔角的BaF2柱体,保证激光通过的区域为高温炉长度为11.43 cm的恒温区域,避免两侧温度梯度对实验测量的影响。BaF2柱体外端嵌入不锈钢盖板中以环氧树脂胶实现粘接,盖板与高温气室主体以氟橡胶O圈实现机械密封,两端的循环水冷系统用于降低高温气室外端温度,保证胶粘密封和O圈密封处的安全性。气室内部压力由精度为0.25 %的压力传感器(Inficon,CDG025D)实时监测。位于高温测量室上方的是放有催化剂(三丰催化剂,S101 中温催化剂)的催化腔,位于其下方的是与催化腔相同尺寸的空腔,催化腔和空腔均一端与高温测量气室连通,一端由不锈钢管连接至高温管式炉外侧并可由针阀控制其开度。
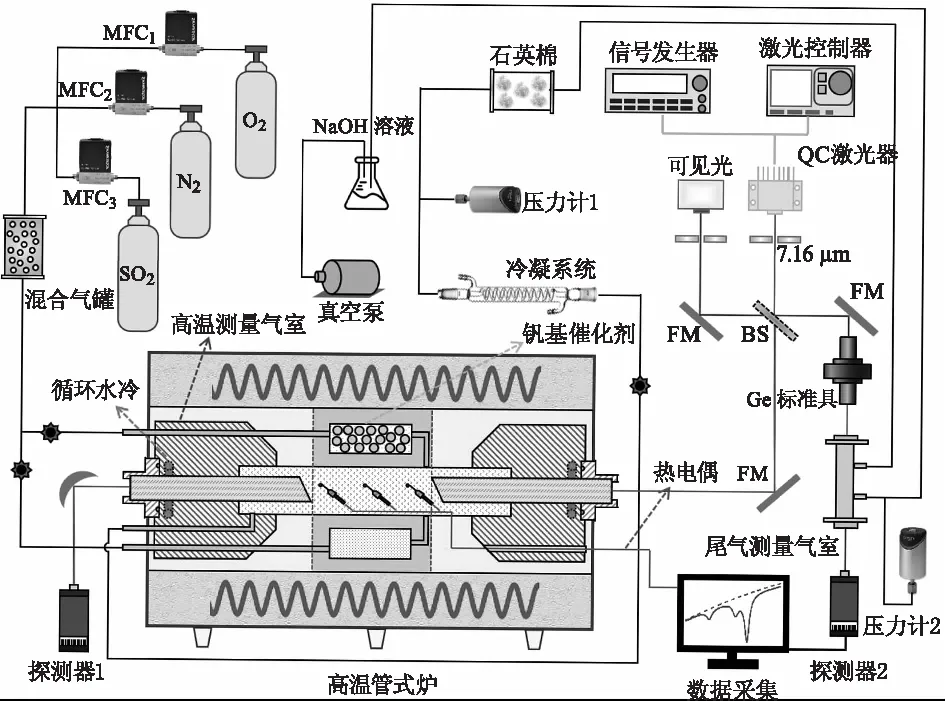
图1 同步测量SO2/SO3实验装置系统(FM—平面反射镜;BS—分束镜)Fig.1 Schematic of the experimental apparatus for simultaneous measurements of SO2 and SO3 (FM—Flat mirror;BS—Beam spliter)
在实验过程中,SO2,N2和O2的流量分别由质量流量计(MFC,Sevenstar,CS200D)控制并完成混合气体配比;流经装满聚四氟小球的玻璃腔体混合均匀;接着经过催化腔完成SO2的催化氧化并生成SO3;SO2和SO3的混合气体在真空泵作用下流经光学高温测量气室完成吸收光谱的测量,接着在高温炉出口下游被冷凝系统冷却,冷凝为液滴的SO3被吸附在装有石英棉的玻璃腔体内,剩余的SO2流入尾气测量气室完成浓度测量;最后,SO2由NaOH溶液吸收并由真空泵将尾气排出。
本文使用7.16 μm的量子级联激光器(HAMAMATSU,LE0981QCL)测量高分辨率SO2和SO3光谱及其浓度。这支QCL激光器可调谐范围为1393~1399 cm-1且在1397 cm-1处的输出功率为50 mW,工作电流和温度分别为0.84 A和24.5 ℃。激光器温度和电流分别由温度控制器(TC10 LAB,Wavelength)和电流控制器(QCL1500 LAB,Wavelength)实现调节。实验中采用信号发生器(Keysight,33510B)产生频率为1 kHz的锯齿波来调制该QCL用于扫描SO2和SO3的光谱。激光光束被ZnSe分束镜分为两束,一束射入高温测量气室,另一束射入尾气测量气室。两束出射光在被探测器(Vigo System PVI-4TE-10.6)接收之前先通过宽带滤光片(Spectrogon,NB-7230-180 nm)以减小热辐射对吸收信号带来的影响。自由光谱区为0.0164 cm-1的Ge标准具用于时域频域转换。图1中的可见光用于辅助调节光路。
本文中的实验温度范围为500~1000 K,在每个实验温度点都按照如下步骤进行操作:①催化腔上游阀门关闭,空腔上游阀门开启,测量反应前SO2吸收光谱在该温度下随压力(3~20 kPa)的变化关系。②空腔上游阀门关闭,催化腔上游阀门开启,部分SO2催化氧化为SO3,在高温气室中测量得到SO2和SO3混合气体的光谱,在尾气气室中测量得到剩余SO2常温光谱。③空腔和催化腔上游阀门均关闭并切断气源。SO2被很快抽干但SO3由于不断从催化剂表面解析,因此短时间内依然能观测到。此时即可采集得到无SO2干扰的SO3的吸收信号。④最后,采用干燥空气将整个实验管路吹扫3~5 h以便去除吸附在管道壁面及催化剂表面的H2O,SO2和SO3。当吹扫至无吸收时采集背景信号。
4 实验结果分析
SO2气体的分子结构与H2O相似,其自身碰撞展宽系数通常是空气碰撞展宽的3~5倍。由于本文实验中配比的SO2浓度为2.5 %~5 %,根据公式(5),如果未考虑自身碰撞展宽会导致最大为20 %的误差。另外,对于本文实验测量波长内的SO2谱线,HITRAN数据库[32]中提供的的SO2自身展宽系数不确定度为20 %或估计值,因此有必要在100 %浓度SO2中测量SO2的自身碰撞展宽系数与谱线线强。图2展示了常温不同压力(0.5~80 kPa)条件下纯SO2气体在1397 cm-1附近的吸收光强It,背景信号I0和Ge标准具干涉信号。
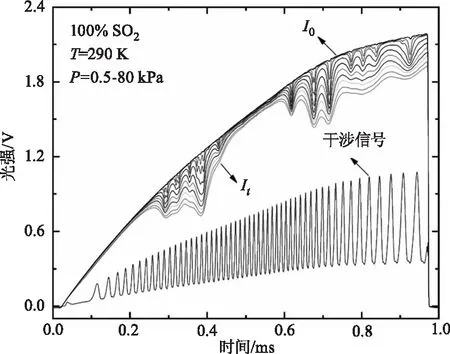
图2 常温条件下不同压力SO2光谱测量信号(包括吸收信号It,背景信号I0和Ge标准具干涉信号)Fig.2 Measured absorption intensity It,background signial I0 and Ge etalon signal at the temperature of 290 K and pressures of 0.5~80 kPa in pure SO2 gas
SO2自身光谱参数标定完成后,开始对配比之后的混合气体SO2(2.5 %)/N2(47.5 %)/O2(50 %)中SO2的吸收率进行标定,此处选用SO2在该测量波段内的吸收峰值(1397.16 cm-1)用于标定SO2在混合气体中的吸收率随温度压力的变化关系。图3(a)展示了SO2吸收峰值在不同温度工况下随压力的变化关系,在每个温度点SO2吸收峰值均随温度升高呈接近线性增大关系,弱非线性主要由于谱线展宽随压力升高而增大导致吸收随压力变化非完全严格线性关系。图3(b)展示了SO2吸收峰值在不同压力工况下随温度的变化关系,在每个压力工况下SO2吸收随温度升高而减小,且变化率随温度升高而增大。
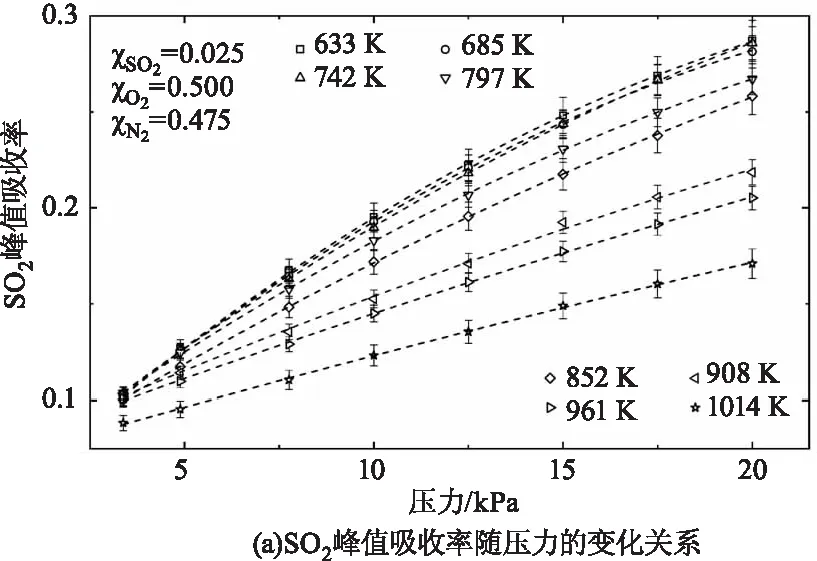

图3 SO2在波数为1397.16 cm-1处的峰值吸收率随压力和温度的变化关系Fig.3 The dependence of the SO2 peak absorbance at 1397.16 cm-1 on temperature and pressure
SO2光谱参数测量与峰值吸收率标定完成后,开始高温同步制备和测量SO3实验。本文实验中高温测量气室温度由高温管式炉实现精准控制,并以50 K的温度梯度由500 K上升至1000 K。当温度低于600 K时,混合气体经过催化腔时并未观察到SO3的吸收光谱,说明低温条件下(< 600 K)SO2未能催化氧化为SO3。虽然未发生催化氧化反应,但在催化腔中却明显观测到了SO2在钒基催化剂表面的吸附效应。图4对比展示了在温度为590 K,压力为30 kPa的条件下,4 %的SO2与O2,N2的混合气体分别通过空腔和催化腔时的SO2光谱吸收率大小。其中,虚线是实验测量数据,实线为空腔和催化腔内测得SO2吸收率的拟合数据,拟合残差均小于峰值吸收率的2 %。从两者吸收峰值处明显看出,当SO2混合气体流经催化腔时部分SO2被吸附于催化剂表面,导致SO2浓度减小,进而使得测量的SO2吸收光谱变弱。
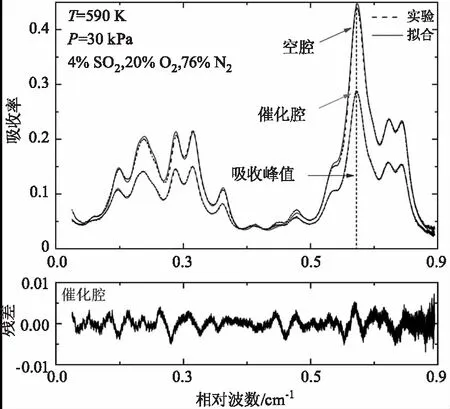
图4 温度590 K,压力30 kPa条件下4 % SO2的混合气体分别流经空腔和催化腔时测量得到的1397cm-1附近的SO2吸收光谱Fig.4 The measured SO2 spectra at around 1397cm-1 when the gas mixtures flowing through the empty and catalytic cavities,respectively at T=590 K and P=30 kPa with a SO2 concentration of 4 %
图5展示了温度为590 K时不同压力下(0.6~30 kPa)测量得到的SO2在钒基催化剂表面的吸附率,其中吸附率φ的表达式如下:
(6)
由图5可知,SO2吸附率随压力呈接近线性上升关系,即压力越大,SO2在催化剂表面的吸附性就越强,且在更低温度条件且未发生催化反应时依然观测到了相同的SO2吸附性随压力变化关系。然而当温度高于600 K时,由于吸附于催化剂表面的SO2开始催化氧化成SO3,因此在该高温下无法定量得到SO2的吸附性特性。
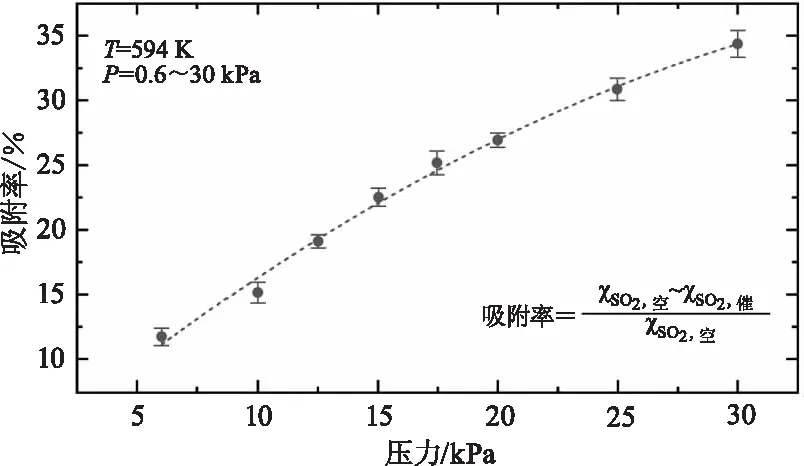
图5 温度为590 K条件下,SO2气体在中温钒基催化剂(S-101)表面的吸附率(%)随压力(0.6~30 kPa)的变化关系Fig.5 The pressure dependence of the SO2 adsorption on the surface of the vanadium catalysts at T=590 K
当高温测量气室温度加热至高于600 K时,开始观测到SO3光谱的出现,此时SO2开始在催化剂表面与O2反应生成SO3。为了将测量得到的SO2/SO3混合吸收光谱解耦分别得到各自光谱,进而通过Beer-Lambert定律得到两种气体的浓度,首先需要得到SO2和SO3的独立光谱结构和谱线参数。SO2的光谱数据(如谱线中心波数、线强、碰撞展宽系数等)在HITRAN数据库中可以查阅,且在本文中也通过测量100 % SO2气体对数据库数据进行了验证和修正。对于SO3的光谱参数和结构,HITRAN等国内外光谱数据库仅提供常温条件下的光谱数据,然而高温SO3的光谱结构比常温条件下复杂的多,常温数据无法参考,因此需要获取高温条件下的SO3独立光谱结构。由于SO3较为活泼的物理化学性质,其标准气体难以制备,因此本文通过实验步骤③得到高温SO3独立光谱。如图6所示,点线为742 K,0.75 kPa条件下在步骤③中测量得到的SO3吸收光谱,竖直线表示HITRAN数据库提供的该波长范围的SO3谱线位置,其长度表示了相对线强,实线为通过HITRAN数据库提供谱线及其光谱参数的理论计算结果。由图6可以看出,实际测量得到的高温SO3光谱与HITRAN数据库理论计算光谱存在很大差异,高温SO3测量光谱的谱线数量远多于数据库提供谱线数量。由于SO3高温独立光谱在低压(<1 kPa)条件下获得,在该压力下谱线展宽窄重叠较弱,可以观测到细致的光谱结构,因此本文将SO3测量光谱中的吸收峰位置作为各条谱线的中心波长,采用Voigt线性模型(如公式(3)所示)描述谱线线型,通过Levenberg-Marquardt非线性拟合方法对实验测量高温SO3吸收率进行拟合,得到各谱线的线强,展宽等光谱参数。由于该波长范围内绝大部分可观测到的SO3谱线在同一振转谱带中,其碰撞展宽系数相近,且拟合过程中,为减小拟合自由度防止过拟合,因此所有SO3谱线的碰撞展宽系数设定为同一个值。
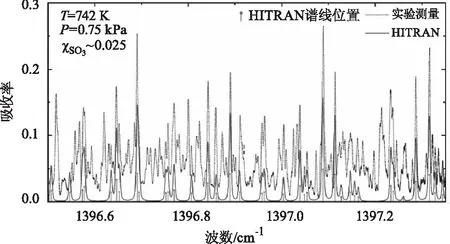
图6 温度为742 K,压力为0.75 kPa的条件下测量得到的SO3独立光谱与HITRAN数据库数据理论计算结果对比Fig.6 Comparison between the measured high-temperature SO3 spectra and the HITRAN simulation at T=742 K and P=0.75 kPa
高温SO3光谱结构和模型参数确定后,即可对催化氧化反应中测量得到的SO2/SO3耦合光谱进行解耦。以SO2和SO3的光谱模型参数和测量的无吸收基线为基础,对采集到的SO2/SO3混合气体的吸收光强进行同步拟合,同时得到SO2和SO3的吸收率,从而计算得到两种气体的浓度,进而计算得到该温度和压力下的反应转化率。图7展示了温度为908 K时测量得到的不同压力(7~20 kPa)条件下SO2/SO3耦合光谱及同步拟合解耦后得到的两者各自吸收率。点线和点划线分别代表解耦后SO2和SO3的独立光谱,两者组成的混合光谱和实验测量光谱的误差在不同工况下均在10 %以内,说明了本文SO3高温光谱模型的准确性和同步拟合的可靠性。
在图7的温度压力条件下,SO3光谱接近连续谱,而SO2光谱相对稀疏,在测量波长范围的中间和两侧吸收很弱接近未吸收区。因此SO3的测量波长选择在SO2吸收非常弱的测量范围两侧,而SO2的测量波长选择在其吸收最强的峰值处。由图7可以看出,在同一温度下,压力越高,SO2的吸收越弱,SO3的吸收越强,说明SO2催化氧化为SO3的转化率随压力上升而增大。
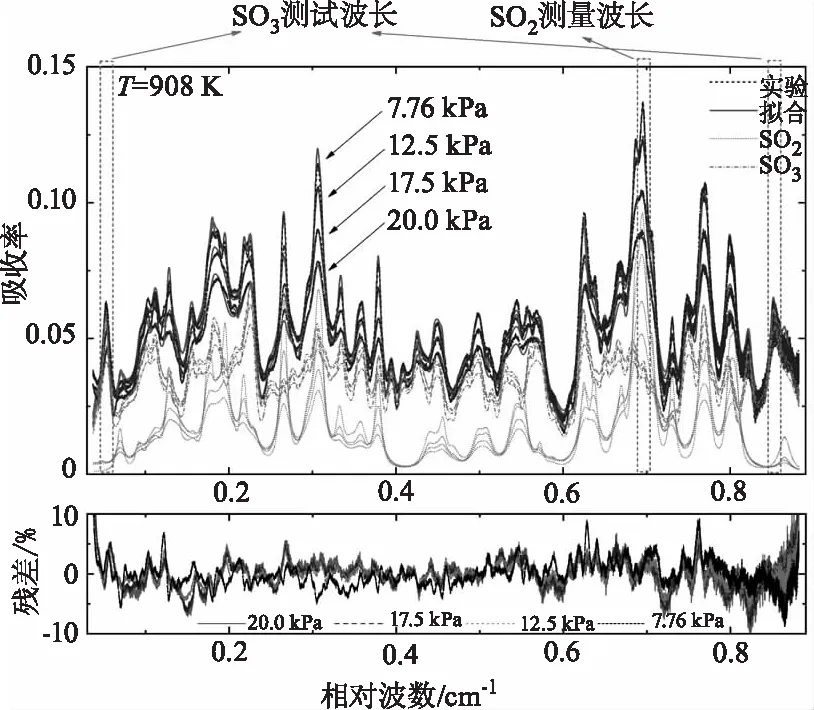
图7 温度为908 K条件下在SO2催化氧化反应过程中测量得到的不同压力(7~20 kPa)下的SO2/SO3混合气体光谱以及拟合残差Fig.7 The measured spectra of the SO2/SO3 gas mixtures and the best-fit residuals during the catalytic oxidation of SO2 at T=908 K and P=7~20 kPa
为了进一步探究SO2催化氧化为SO3转化率随温度和压力的变化关系,本文继续开展了温度范围为600~1000 K,压力范围为3~20 kPa的实验探究,在该温度范围内均可观察到SO2催化氧化反应的发生。各个温度压力工况的SO2催化氧化反应转化率η可由公式(7)计算得到:

(7)
根据硫元素守恒,反应后生成的SO3摩尔数应等于反应中消耗的SO2摩尔数。由于SO3气体浓度的测量精度低于SO2,因此本文采用SO2气体反应前后的浓度差来计算该催化氧化反应的转化率。图8展示了测量得到的不同温度压力工况下SO2催化氧化为SO3的转化率,图8(a)中不同形状的点代表的是各个测量温度下的反应转化率随压力的变化趋势,虚线根据同形状的测量数据由多项式拟合得到。可以看出,各个温度下反应转化率随压力上升均呈现增大趋势,且温度越高,转化率随压力的变化越明显。并且,每个温度曲线的增长率随压力上升而减小,即压力越大,转化率对压力的变化越不敏感。图8(b)展示的是同一压力工况下反应转化率随温度的变化关系。在测量的各个压力工况下,反应转化率随温度上升均呈现先增长后减小的变化趋势。并且,压力越小转化率随温度的变化幅度越大,对温度变化越敏感。不同压力下转化率最高时对应的温度点均在750~800 K之间,这与该中温钒基催化剂(S-101)的工业使用最佳温度约为500 ℃(773 K)相吻合,进一步证明了实验测量的准确性与可靠性。压力为3.38 kPa、4.89kPa和7.76 kPa的转化率曲线在800 K和850 K两个温度点处的不确定度较大,主要因为低压条件下SO2和SO3的吸收较弱,实验信噪比较低,且存在低压条件下少量未清除干净仍吸附在催化剂表面的SO2和SO3气体析出,进而对实验测量结果造成影响。
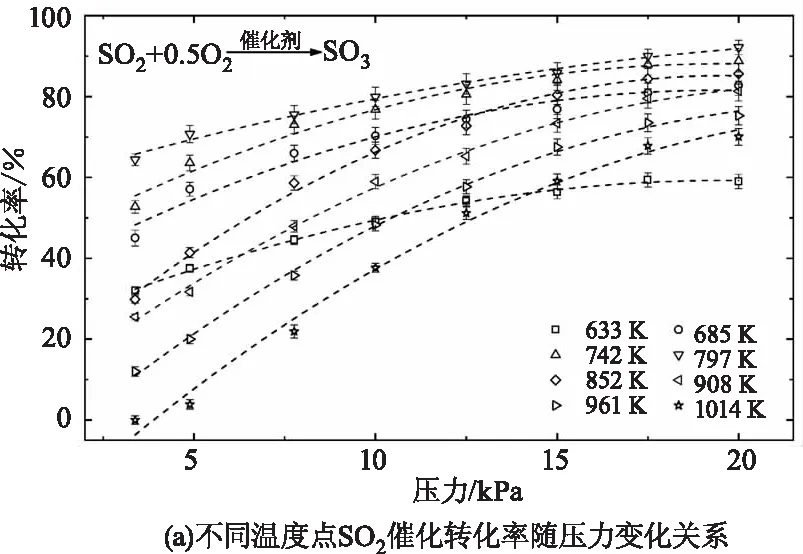
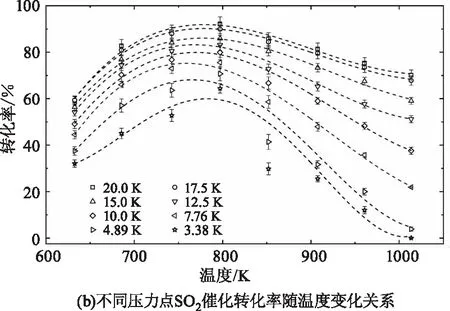
图8 SO2催化氧化转化率随压力(3~20 kPa)和温度(600~1000 K)的变化关系Fig.8 The dependence of the measured SO2 heterogeneous conversions on and pressure(3~20 kPa)and temperature (600~1000 K)
为了验证高温同步测量SO2和SO3浓度的准确性,本文还在高温气室出口下游处设计了冷凝和吸收装置,将混合气体中的SO3脱除,从而在尾气测量腔中单独测量反应后剩余SO2浓度,并与同步测量得到的SO2浓度对比,两者在各种工况下的差别均小于7 %,进一步验证了高温条件下同步测量结果的准确性和可靠性。
5 结 论
本文描述了一套基于QCL中红外激光吸收光谱技术的高温SO2和SO3同步测量实验系统。本文首先测量了实验波长范围内SO2气体谱线的线强与自身碰撞展宽等光谱参数,对HITRAN数据库数据进行了验证和修正,并且标定了SO2的峰值吸收率随温度压力的变化关系。高温加热实验在高温管式炉中进行并实现SO3的同步制备和测量。在温度低于600 K时,SO2的催化氧化反应未能发生,但观测到SO2在催化剂表面有明显的吸附效应,并定量测量得到SO2吸附率随压力上升而增大。在温度高于600 K时,SO2和O2在钒基催化剂的作用下反应生成SO3,通过完善高温SO3光谱模型及其参数,实现了在反应过程的混合气体中SO2和SO3的同步测量并得到其各自光谱结构和浓度,进而计算得到SO2催化氧化反应的转化率及其随温度压力的变化关系。在同一温度下,SO2转化率随压力上升呈对数上升趋势;在压力不变的条件下,SO2转化率随温度上升先增大后减小,在温度为750~800 K范围内转化率达到最大值。
猜你喜欢
中国科技财富(2022年8期)2022-12-18
昆钢科技(2022年2期)2022-07-08
物理学报(2022年10期)2022-06-04
天津诗人(2021年1期)2021-11-12
昆钢科技(2021年4期)2021-11-06
物理实验(2020年12期)2021-01-06
科学技术与工程(2020年4期)2020-04-10
分析化学(2018年12期)2018-01-22
中学化学(2017年5期)2017-07-07
未来英才(2016年13期)2017-01-13
