
First reference genome assembly of the Indochinese silvered langur (Trachypithecus germaini)
2022-08-05 10:42Jin-WeiWu,RuZhang,Ya-LinQin等
Zoological Research 2022年4期
DEAR EDITOR,
Of the seven genera recognized in Asian colobines,Trachypithecusis the only genus that contains species groups.Compared with the species groups characterized by calcium tolerance (T.francoisispecies group),multi-male,multi-female society (T.obscurusspecies group),and impressive hybridization (T.pileatusspecies group),theT.cristatusspecies group is distinguished by its southernmost distribution and silvery appearance.Hence,Trachypithecusis an excellent model for investigating evolutionary radiation and behavioral adaptation in Asian primates.However,comprehensive comparison of species groups remains difficult due to the lack of a reference genome for theT.cristatusspecies group.In the current study,based on Nanopore sequencing,we produced a high-qualityde novoassembly of the Indochinese silvered langur (Trachypithecus germaini)genome as a representative of theT.cristatusspecies group.The assembled genome was 2.91 Gb in size,with a contig N50 of 55.90 Mb.The genome was predicted to contain 20 332 protein-coding genes,and genome synteny analysis betweenT.germainiandT.francoisiindicated a good collinear relationship.Demographic history analysis indicated that theT.germainipopulation declined during glacial periods,possibly due to climate change and human activity.The highquality genome of the Indochinese silvered langur should provide a valuable resource for a deeper understanding of the natural history and social evolution ofTrachypithecusspp.,as well as adaptive radiation in primates.
The Indochinese silvered langur (T.germaini),also known as the Indochinese Lutung,is distributed in Thailand,Burma,Cambodia,Laos,and Vietnam (Figure 1A).As a colobine primate,this species lives in typical one-male,multi-female units,and displays territory defense against other groups(Rowe &Myers,2016).Like other colobines,T.germainilangurs are well-adapted to their high-fiber folivorous diet,with developed bilophodont molars (Wright &Willis,2012) and enlarged,sacculated ruminant-like stomachs containing bacteria for cellulose fermentation (Davies &Oates,1994).
TheTrachypithecusgenus is comprised of four species groups (Roos et al.,2020).TheT.francoisispecies group is restricted to karst habitats in Laos,Vietnam,and southwestern China (Figure 1A).Individuals live in one-male,multi-female units and have adapted to high calcium ion concentrations in their blood (Liu et al.,2020).TheT.obscurusspecies group is mainly distributed in the mountainous forests of southwestern China and the Indochinese Peninsula (Figure 1A).Different from all otherTrachypithecusspp.,those in theT.obscurusgroup (e.g.,T.crepusculus) are organized in multi-male,multifemale social units (Xiong et al.,2017).TheT.pileatusspecies group is distributed in northeastern India,Bhutan,eastern and central Bangladesh,northwestern Myanmar,and southwestern China,showing mixed distribution withSemnopithecus.Thus,members in theT.pileatusspecies group exhibit morphological characteristics of bothTrachypithecusandSemnopithecus(Figure 1A) (Osterholz et al.,2008;Wang et al.,2015),suggesting incomplete lineage sorting or hybridization events during speciation.In contrast,members of theT.cristatusspecies group can be distinguished based on their southernmost distribution,restricted to the rainforests of Peninsular Malaysia,and their unique gray and silver pelage (Rowe &Myers,2016)(Figure 1A).
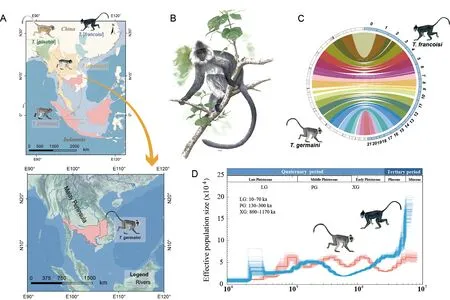
Figure 1 Geographical distribution and genomic analysis of T.germaini
As the only genus to contain species groups in the subfamily Colobinae,species withinTrachypithecushave evolved distinct morphological,physiological,social,and behavioral traits.Thus,this genus represents an excellent model for studying theories of primate evolution and the underlying genetic mechanisms related to diversification,speciation,hybridization,introgression,adaptive evolution,and social differentiation.Therefore,it is necessary to obtain a set of genomes covering all four species groups.Currently,only theT.francoisispecies group reference genome has been reported (Liu et al.,2020),with theT.obscurusandT.pileatusspecies group genomes completed and in the process of being published.However,aT.cristatusspecies group reference genome is still lacking.
To establish a reference genome forT.germaini(Figure 1B),we collected blood from a maleT.germainilangur in Nanning Zoo (Nanning,Guangxi Province,China).Genomic DNA was extracted using a QIAGEN Blood &Cell Culture DNA Mini Kit (QIAGEN,Germany).A short-insert-size library was constructed and sequenced using the MGISEQ-2000 platform (BGI,China).Nanopore libraries were prepared using the BluePippin system (Sage Science,USA) and sequenced using the PromethION platform (Oxford,UK).
After checking quality,208.05 Gb of clean short reads were acquired and used to estimate genome size byK-meranalysis(Supplementary Tables S1,S2).The estimated genome size was~3.05 Gb,with a heterozygosity ratio of 0.21%(Supplementary Figure S1 and Table S2).The 349.10 Gb of Nanopore reads were used to performde novoassembly.The initial genome was generated using NextDenovo v2.31 and polished by NextPolish v1.3.1,with both short and long reads.To further improve genome assembly,two rounds of polishing were executed with Pilon v1.23 using short reads.The final assembled genome was 2.91 Gb,with a contig N50 of 55.90 Mb (Supplementary Table S3).
The clean short reads and Benchmarking Universal Single-Copy Orthologs (BUSCOs) were used to evaluate genome assembly and completeness in gene regions,respectively.All clean short reads were mapped to the assembled genome using Burrows-Wheeler aligner (BWA) v0.7.15 with default settings (Supplementary Table S4).To assess completeness,we used the 4 104 BUSCOs from the mammalia_odb9 dataset to align the assembly using BUSCO v3.0.2.Results showed 94.7% complete BUSCOs (Supplementary Table S5),which is superior to that inT.francoisi(Liu et al.,2020).In addition,whole-genome synteny was performed between theT.germainiandT.francoisigenomes using LASTZ v1.04.03.Results showed thatT.germainihad a high conserved synteny withT.francoisi(Figure 1C).Overall,the assembled genome demonstrated high integrity and continuity.
De novoand homology-based approaches were applied to predict the repeated sequences of transposable elements(TEs) and tandem repeats in theT.germainigenome.Novel TEs were identified and classified using RepeatModeler.Known TEs at the DNA and protein level were detected using a homology-based approach in RepeatMasker and RepeatProteinMask.Tandem Repeat Finder was used to identify tandem repeats.In total,1.44 Gb of repeat sequences were identified,accounting for 49.43% of the assembled genome,which is comparable to that of other primates(Supplementary Tables S3,S6).In total,1.38 Gb of TEs were predicted,accounting for 47.58% of the assembled genome.Among TEs,long interspersed nuclear elements (LINEs) were most abundant (22.99% of the assembled genome),followed by short interspersed nuclear elements (SINEs;13.74%) and long terminal repeat (LTR) retrotransposons (7.68%)(Supplementary Table S7).
Both homology-andde novo-based prediction methods were used to predict gene models of the repeat-masked genome using EvidenceModeler (EVM).For homology-based prediction,protein sequences ofT.francoisi,Rhinopithecus roxellana,Macaca mulatta,andHomo sapienswere aligned against theT.germainigenome using TBLASTN (E-value=1e-5).Solar software was used to conjoin BLAST hits and GeneWise was applied to predict gene structures.Forde novo-based prediction,AUGUSTUS and GENSCAN were used to predict coding genes.A non-redundant gene set was generated based on gene models from EVM,which predicted 20 332 protein-coding genes in theT.germainigenome(Supplementary Table S8).To evaluate the quality of the predicted genes,we compared gene features,including distribution of mRNA length,CDS length,and exon length,inT.germainiwith other primates,which indicated a similar distribution pattern (Supplementary Figure S2).Completeness of the annotated genes was assessed using BUSCO v3.0.2 with default parameters,which detected 93.9% complete BUSCOs (Supplementary Table S9).These findings indicated the presence of high-confidence gene models.
Functional annotation of the predicted genes was performed by alignment to the SwissProt,TrEMBL,and NR databases using BLASTP.For the prediction of structural domains and motifs,the predicted genes were searched against the SMART,ProDom,Pfam,PRINTS,PROSITE,and PANTHER databases using InterProScan v5.25.In total,16 756 proteincoding genes were functionally annotated,accounting for 82.41% of the predicted genes (Supplementary Table S10).
Non-coding RNA genes include highly abundant and functionally important RNAs such as transfer RNAs (tRNAs),ribosomal RNAs (rRNAs),small nuclear RNAs (snRNAs),and microRNAs (miRNAs).Here,the tRNA genes were searched using tRNAscan-SE.The rRNA genes were predicted by alignment to the Vertebrate rRNA Database using BLASTN(E-value=1e-5).The snRNA and miRNA genes were predicted using INFERNAL against the Rfam database with default parameters.In total,547 miRNAs,365 tRNAs,237 rRNAs,and 2 095 snRNAs were identified (Supplementary Table S11).
The demographic history ofT.germainiwas inferred using the Pairwise Sequentially Markovian Coalescent (PSMC)approach based on single nucleotide polymorphisms (SNPs).Candidate SNPs were identified using SAMtools and BCFtools v1.9.The candidate SNPs were then filtered if their depth of coverage less than a third or greater than twice the average depth.The PSMC analysis was performed to infer effective population size.Results indicated that population decrease inT.germainiwas consistent with the Xixiabangma Glaciation(XG,1 170–800 thousand years ago (ka)) and Last Glacial Maximum (LGM,70–10 ka),similar to that reported forT.francoisi(Liu et al.,2020) (Figure 1D).The climatic shifts during the glacial and interglacial periods and the emergence ofHomo sapiensin the Late Pleistocene may be associated with the decline in theT.germainipopulation.
In this study,we sequenced and assembled a high-quality reference genome ofT.germaini.As the first reference genome of theT.cristatusspecies group,this study has important implications for further studies of the natural history,social evolution,adaptation radiation,and species conservation ofTrachypithecusand Asian colobines.
DATA AVAILABILITY
All raw sequencing reads and the genome assembly were deposited in the National Center for Biotechnology Information(NCBI) database (BioProjectID PRJNA822022).Genome assembly was deposited in Science Data Bank(http://www.scidb.cn/cstr/31253.11.sciencedb.j00139.00009)and GSA under accession No.GWHBJLC00000000.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
X.G.Q.designed the study.H.L.S.,S.W.,and N.H.collected the samples.X.G.Q.,C.Z.,and W.J.W.performed genome sequencing.R.Z.performed data analysis.R.Z.,W.J.W.,and X.G.Q.wrote the paper.All authors added materials and read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Nanning Zoo for support and permission to perform blood sampling of the study animals.
Jin-Wei Wu1,#,Ru Zhang1,#,Ya-Lin Qin1,Li Wang1,Ming-Bo Han1,Zi-Fan Zhao1,Pei Jing1,Hai-Lu Sun2,Song Wang3,Ning Huang3,Bao-Guo Li1,Dong-Dong Wu4,Chi Zhang5,*,Xiao-Guang Qi1,*
1College of Life Sciences,Northwest University,Xi’an,Shaanxi710069,China2BGI-Shenzhen,Shenzhen,Guangdong518083,China
3Nanning Zoo,Nanning,Guangxi450107,China
4Kunming Institute of Zoology,Chinese Academy of Sciences,Kunming,Yunnan650223,China
5Key Laboratory of Adaptation and Evolution of Plateau Biota,Northwest Institute of Plateau Biology,Chinese Academy of Sciences,Xining,Qinghai810008,China
#Authors contributed equally to this work
*Corresponding authors,E-mail:qixg@nwu.edu.cn;zhangchi2@bgi.com

- Zoological Research的其它文章
- Zoological Research call for papers of Cavefish Special Issue
- Fuel source shift or cost reduction:Context-dependent adaptation strategies in closely related Neodon fuscus and Lasiopodomys brandtii against hypoxia
- Ecological study of cave nectar bats reveals low risk of direct transmission of bat viruses to humans
- Population and conservation status of a transboundary group of black snub-nosed monkeys (Rhinopithecus strykeri) between China and Myanmar
- Nucleus accumbens-linked executive control networks mediating reversal learning in tree shrew brain
- Europe vs.China:Pholcus (Araneae,Pholcidae) from Yanshan-Taihang Mountains confirms uneven distribution of spiders in Eurasia