
Allele-specific expression analyses reveal immune divergences between ibex and goat species
2022-08-05 10:42Zhi-RuiYang,Jia-XinLi,Zhu-QingZheng等
Zoological Research 2022年4期
DEAR EDITOR,
Understanding the different immune responses of wild and domestic caprid species is critical for addressing certain zoonotic diseases.In this study,we generated blood transcriptomes of 13 Siberian ibex and domestic goat hybrids and performed allele-specific expression and splicing analyses.Results showed that genes exhibiting significant imbalance between the ibex and goat were highly related to the Toll-like receptor (TLR),antigen recognition,and immune activation pathways.Comparative genomic analysis of the species revealed that immune function-related genes were under strong selection pressure in the domestic goat.These allelic imbalances in gene expression may be related to fixed divergent sites incis-regulatory elements,which may affect the expression of immune-related genes and contribute to the adaptive diversity between ibex and goat species.
The Siberian ibex is a wild relative of the domestic goat and is distributed in the mountains of Central and East Asia.It is classified as a near-threatened species in the IUCN Red List(https://www.iucnredlist.org) and is considered an endangered species in the China Red Data Book of Endangered Animals.Siberian ibex and domestic goats,which are able to produce fertile offspring,often cohabitate in the same ecosystem and share multiple pathogens,such as petits ruminants virus(Benfield et al.,2021).Understanding the differences between these wild and domestic species is critical to the safety of livestock and wildlife.Hybrid offspring between the ibex and goat provide an ideal model for transcriptional comparison between the species.In ibex-goat hybrids,allele-specific expression can directly reflect gene expression divergence in parental species,and indirectly assess the function of variations incis-regulatory elements (Wittkopp et al.,2004).
In this study,we collected peripheral blood samples from 13 adult female ibex-goat hybrids (Figure 1A).Using a bin map of the whole genome,we divided the 13 hybrids into three F1 and 10 F2 samples (Supplementary Methods).On average,for each sample,11X re-sequencing data and 3.5 Gb of transcriptome data were generated,respectively.Ibex and goat genomes were downloaded for genomic comparison.
We first performed genomic comparison between the ibex and goat to provide insight into the effects of natural selection on these divergent species.We detected signals of positively selected genes (PSGs) and evaluated the number of substitutions per synonymous (Ks) and nonsynonymous (Ka)sites for both species based on their genomes.We identified 96 and 74 PSGs in the goat and ibex lineage,respectively(Supplementary Tables S1–S6).Pathway enrichment analysis indicated that the 96 goat PSGs were significantly enriched in two pathways (correctedP-value<0.05),i.e.,immune system and olfactory signaling pathways.In contrast,the 74 ibex PSGs were enriched in 13 pathways (Figure 1B).Comparison of theKa/Ks ratios for each Gene Ontology (GO) category revealed that the elevated pairwiseKa/Ks ratios were enriched in immune-defense of domestic goats (Supplementary Figures S1–S3 and Table S7).
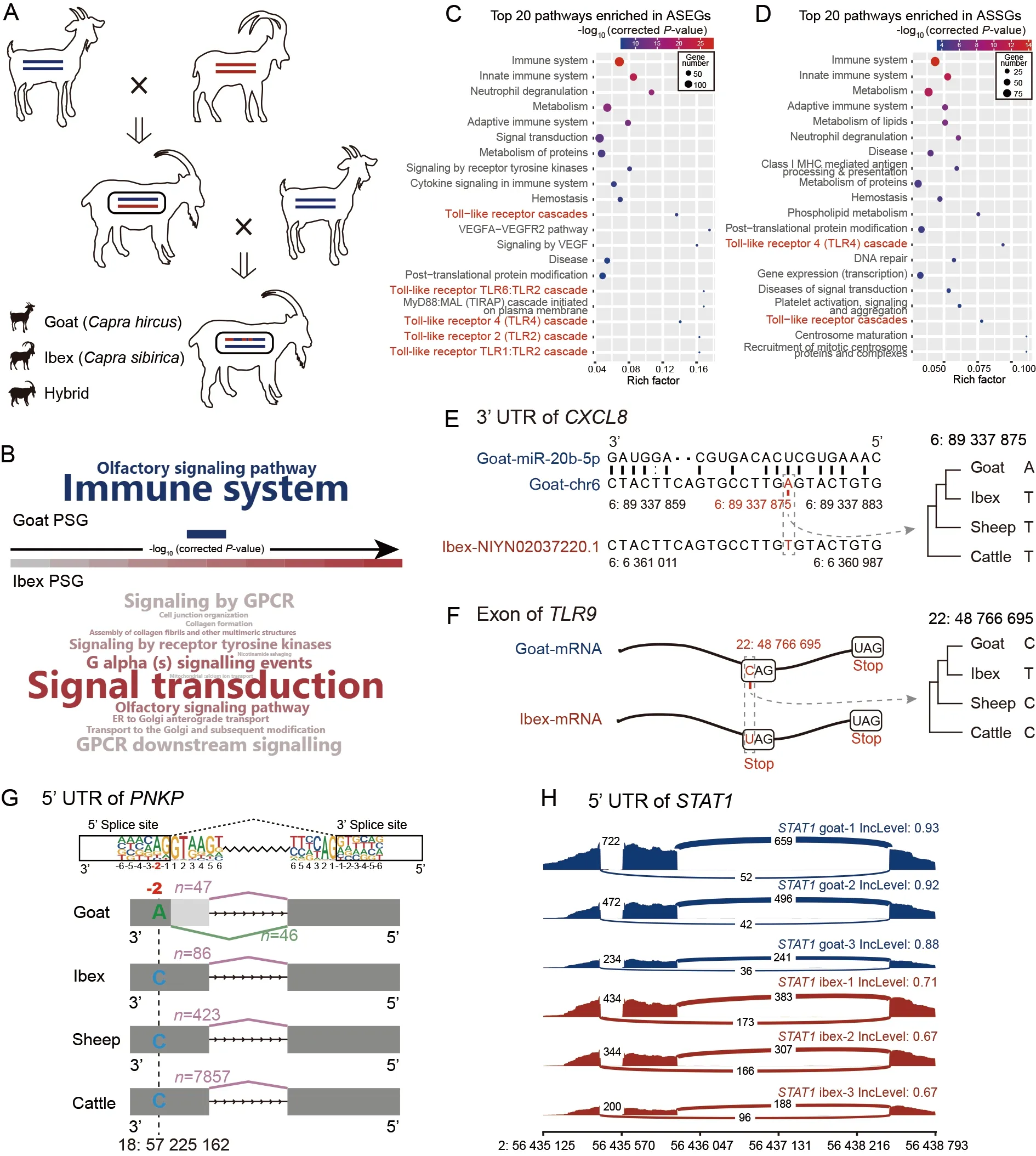
Figure 1 Allelic imbalance expression and genomic divergence between ibex and goat
We next performed transcriptome analysis.To identify alleles between goat and ibex hybrids and reduce mapping bias,we first obtained~5 Mb of fixed divergent sites (FDSs)between the two species (Supplementary Methods and Figure S4).A pseudogenome was then constructed by replacing the FDSs from the goat allele to the ibex allele (Supplementary Methods and Figure S5).After mapping to the goat genome and pseudogenome,the results were merged for allelic expression and splicing analyses.The expression ratios of goat and ibex alleles in the FDSs were calculated.The FDSs with imbalanced expression were filtered as allele-specific sites.Based on these allele-specific FDSs,we used a bin map to distinguish the ibex and goat fragments in the 13 hybrids for further analysis (Supplementary Methods and Supplementary Figure S6).A total of 896 genes with at least three allelespecific sites were regarded as allele-specific expression genes (ASEGs) (Supplementary Figure S7),which included 853 protein-coding genes,39 non-coding RNAs,and four pseudogenes.Allele-specific splicing genes (ASSGs) were detected in three F1 hybrids.Transcriptome reads were divided into two genetic alleles.Splicing events were detected by rMATS v4.0.1 and tested using the likelihood-ratio method.In total,827 statistically significant differential splicing events were found (Supplementary Methods and Figure S8).
Enrichment analyses of ASEGs and ASSGs revealed significant associations with immune system-related pathways(Figure 1C,D;Supplementary Tables S8,S9),consistent with the goat PSG-enriched immune system pathway.We further explored the intersections between ASEGs/ASSGs and published immune-related signaling pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG).For the ASEGs,TLR,a pattern recognition receptor (PRR),exhibited the highest intersections.For the ASSGs,RIG-I-like receptor cascade,another PRR,exhibited the highest intersections,followed by the TLR cascade (Supplementary Figure S9).
The ASEGs in the TLR and immune-related pathways were analyzed in detail (Supplementary Table S10).CXCL8showed high ibex-preferred expression (Supplementary Figure S10A).One FDS in the 3' untranslated region (UTR) had a Tto-A mutation in goats (Figure 1E),which differs from that found in other bovine genomes (Fu et al.,2021).This mutation may increase the binding of mir20b-5p to the 3'UTR ofCXCL8(Figure 1E) (Agarwal et al.,2015).For theTLR9gene,allele expression was lower in the ibex (Supplementary Figure S10B) and contained a C-to-T mutation,introducing a premature stop codon (Figure 1F) associated with nonsensemediated mRNA decay (Lykke-Andersen &Jensen,2015).
Among the identified ASSGs,PNKPplays an important role in DNA repair.It contained one fixed mutation at the -2 site in the 5' UTR,which showed high conservation among individuals (Figure 1G).This mutation (from G in Bovidae to T in goat) and splicing event are both novel in the goat genome(Figure 1G;Supplementary Figure S11).TheSTAT1gene showed one splicing variant,which generated an extended 5’isoform with a nuclear localization signal (Figure 1H;Supplementary Figure S12).This is a newly evolved isoform,conserved in multiple organisms (Supplementary Figures S13–S14).
We combined comparative genetics and functional allelespecific expression to provide multi-omics analysis of adaptive evolution in caprid species.Genomic analysis indicated that immune-related genes were under higher selection pressure in the domestic goat than in the ibex lineage.Transcriptome analysis indicated that the ASEGs and ASSGs in the ibex-goat hybrids were also enriched in immune-related pathways.Thus,these results revealed the importance of immunity in the adaptive evolution of caprine,consistent with enhanced resistance of gastrointestinal nematodes (Zheng et al.,2020)and the brucellosis transmission model in early goat domestication (Fournié et al.,2017).We identified several immune-related pathways with high enrichment factors,such as the TLR cascade,which participates in pathogen recognition and immune activation.The effects of admixture and gene expression have also been studied in humans,with the Neanderthal haplotype associated with significantly increased expression ofTLR1(McCoy et al.,2017).In conclusion,we identified several FDSs that likely affect transcriptome expression by regulating gene expression and splicing,which may ultimately influence immune function.These findings should aid further epidemiological studies,especially the transmission of diseases between wild and domestic animals.
DATA AVAILABILITY
All data used in the study are described in the manuscript and/or Supplementary Materials.The re-sequencing and transcriptome data were submitted to the Science Data Bank database (DOI:10.57760/sciencedb.j00139.00004,31253.11.sciencedb.j00139.00004),National Genomics Data Center(GSA:PRJCA009442,PRJCA009461),National Center for Biotechnology Information (NCBI BioProjectID:PRJNA836147,PRJNA836141),and China National GeneBank Database (CNGB:CNP0002452,CNP0002378).
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
X.H.W.,Y.J.,and W.X.Z.designed and supervised the study;Ming Li,X.L.D.,and Y.J.L.collected the hybrid samples;Z.R.Y.and J.X.L.performed the data analysis with contributions from Z.Q.Z.,Ming Li,Y.W.,and C.Z.;Z.R.Y.,X.H.W.,and H.A.N.drafted the manuscript;R.L.,C.N.C.,and Mao Li revised the manuscript.All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We would like to thank Jun-Jie Shao,Fei Wang,and Jia-Qi Fu from the Laboratory for Genomic Big Data for support during this project.We thank Wei Liu from High-Performance Computing (HPC) of Northwest A&F University (NWAFU) for assistance during this project.We thank the Xinjiang Key Laboratory of Animal Products Quality and Safety and Xinjiang Plush Engineering Technology Research Center for support with sample collection.We also thank Linda Liu for assistance with language polishing.
Zhi-Rui Yang1,#,Jia-Xin Li1,#,Zhu-Qing Zheng1,Chen Zhao1,Yu Wang1,Ming Li1,Hojjat Asadollahpour Nanaei1,2,Xue-Lei Dai1,Yun-Jia Li1,Ran Li1,Chun-Na Cao1,Mao Li3,Yu Jiang1,*,Wen-Xin Zheng4,*,Xi-Hong Wang1,*
1Key Laboratory of Animal Genetics,Breeding and Reproduction of Shaanxi Province,College of Animal Science and Technology,Northwest A&F University,Yangling,Shaanxi 712100,China
2Department of Animal Science,Faculty of Agriculture,Shahid Bahonar University of Kerman,Kerman,PB76169-133,Iran
3Tropical Crops Genetic Resources Institute,Chinese Academy of Tropical Agricultural Sciences,Danzhou,Hainan571700,China
4Xinjiang Academy of Animal Sciences,Urumqi,Xinjiang830011,China
#Authors contributed equally to this work
*Corresponding authors,E-mail:yu.jiang@nwafu.edu.cn;zwx2020@126.com;wxh@nwafu.edu.cn

- Zoological Research的其它文章
- Zoological Research call for papers of Cavefish Special Issue
- Fuel source shift or cost reduction:Context-dependent adaptation strategies in closely related Neodon fuscus and Lasiopodomys brandtii against hypoxia
- Ecological study of cave nectar bats reveals low risk of direct transmission of bat viruses to humans
- Population and conservation status of a transboundary group of black snub-nosed monkeys (Rhinopithecus strykeri) between China and Myanmar
- Nucleus accumbens-linked executive control networks mediating reversal learning in tree shrew brain
- Europe vs.China:Pholcus (Araneae,Pholcidae) from Yanshan-Taihang Mountains confirms uneven distribution of spiders in Eurasia