
植原体侵染对桑树叶片转录组的影响
2022-08-10 02:07杨金宏孔卫青
陕西农业科学 2022年6期
杨金宏,孔卫青
(安康学院陕西省蚕桑重点实验室,陕西 安康 725000)
0 引言
桑树黄化型萎缩病(Mulberry Yellow Dwarf Disease,MYDD)是桑树的重大病害,染病桑树枝短叶小,叶片异常黄化,病株逐渐衰竭而枯死,全国范围内的桑园均可受到该病的侵害,常导致大片桑园毁灭,严重制约蚕业生产的健康发展[1]。该病由植原体(Phytoplasma)引起,这是一类重要的原核致病菌,寄主范围广,能够侵染98科1 000多种植物[2],寄生于植物韧皮部,随植物体内营养流进行运动,先下行到根部,再在植物韧皮部筛管内被动上行诱发症状出现,使病株叶片、叶脉和韧皮部部分细胞的细胞核及细胞器结构发生改变,甚至解体[3],危害严重[4,5]。
目前桑树黄花型萎缩病的研究,主要集中在病株同工酶、代谢物、蛋白质组和miRNA的变化的研究。雷国新等研究发现MYDD桑树枝皮的过氧化物同工酶谱带较正常植株有不同程度的加强,且随染病后经过日数的增多而愈发显著[6]。这与过氧化物酶可参与植物的多种代谢途径,被认为与植物抗病抗逆性关系密切一致。韩雪娟等利用质谱(Mass Spectrometry,MS)分析了健康和MYDD植株叶片汁液中的极性代谢物,发现叶片中有65种代谢物的浓度发生了变化,大部分浓度降低[7]。袁传忠分析桑树韧皮部汁液中有56种代谢物的浓度发生变化[8]。冀宪领研究了桑树叶片蛋白质组响应植原体侵染的变化,发现植原体侵染可以引起叶片中包括参与自由基清除、代谢、防御、碳水化合物和能量代谢等方面15种蛋白质的变化[9]。盖英萍等的研究显示,健康和MYDD桑树韧皮部汁液中miRNA的表达存在差异,且鉴定出部分桑树韧皮部汁液响应植原体侵染的miRNAs[10]。
植原体的侵染对桑树代谢物、蛋白质组、miRNAs表达等均有重要影响,植原体侵染与桑树黄花型萎缩病的发生可能涉及到一系列基因的差异表达,影响到多种代谢过程,形成复杂的基因表达和代谢调控网络。转录组(transcriptome)狭义上指某一生理条件下,细胞内所有mRNA的集合,是连接遗传信息与生物功能蛋白质的必然纽带,也是生物体最重要的基因调控方式。基于此,本文对黄花型萎缩病桑树叶片转录组的变化,及相应的代谢途径和基因进行研究,以揭示桑树响应植原体侵染的分子机理研究提供参考。
1 材料与方法
1.1 实验材料
桑树品种为湖桑32,选择经qPCR验证的健康和有明显感病性状的桑树各5株,用花盆种植于组培室中,分别取每株中部叶片3枚,每枚叶片切取0.1 g用液氮速冻,后保存于-80℃冰箱中备用。
1.2 叶片RNA提取、cDNA文库构建和转录组序列测定
均匀混合上述桑树的叶片组织,液氮研磨,参照Invitrogen公司的TRIzol Reagent说明书提取桑树叶片总RNA。使用1.0%琼脂糖凝胶电泳和Nanodrop检测RNA的纯度(D260/D280)和浓度。经检测合格的RNA样品进行文库构建。文库构建方法参照TruseqTMRNA sample prep Kit(Illumina公司)说明书进行,文库构建完成后,由上海美吉生物医药科技有限公司在IlluminaHiseq 2 000测序平台完成2×100 bp的DNA双末端测序。
1.3 测序数据处理与功能注释
碱基读取得到原始序列数据(raw data)后,对其进行质控处理以获得高质量的测序结果(Clean Data),包括去掉含有接头Reads和低质量Reads(质量值Q小于20、含N较多和长度小于20nt)。后用Trinity软件对Clean Data进行序列拼接和ORF预测,得到转录本和ORF序列特征和长度分布,BLAST软件对预测的氨基酸序列和剩余未预测出ORF的序列与NCBI的非冗余蛋白数据库、String、Gene数据库进行比对(E-value:1e-5),对预测ORF进行功能注释。Bowtie软件以Clean Data和川桑MorusnotabilisSchneid基因组为参考,获得序列在参考基因组或基因上的位置信息及测序样品特有的序列特征信息[11]。
后利用Blast2go软件进行GO功能注释,利用BLAST软件与STRING 9.0数据库(http://string-db.org)和KEGG genes数据库(http://www.genome.jp/kegg/genes.html)比对,以所得COG(Clusters of Orthologous Groups of proteins)number进行功能归类[12],以得到的KO编号查找具体的生物学通路,获得基因可能参与的所有生物学通路。最后使用RSEM软件对UniGenes和基因的表达水平进行定量,以FPKM值作为基因表达水平指标。
1.4 差异表达基因筛选与显著性富集分析
使用edgeR软件进行差异表达基因筛选,符合错误发现率(false discovery rate,FDR)小于0.05且差异倍数(fold change)在3倍以上的基因被定义为样品间差异表达基因(differential gene,DEG)[13]。进一步使用goatools软件对对差异基因进行GO功能显著性富集分析[14],使用KOBAS软件对差异表达基因进行通路富集分析,以寻找与研究背景密切关联的生物学过程[15],与植物抗性基因数据库PRGDB(http://prgdb.org)比对[16]。
2 结果与分析
2.1 桑树叶片转录组测序数据结果
对健康(MO)和病树(MV)叶片转录组测序结果表明(表1),2样本分别获得63 484 370和61 812 660条raw reads,质控处理后,分别获得2样本的测序数据5.87Gb和5.70Gb数据,含61 230 412和59 551 764个reads片段,Q30碱基百分比分别为91.69%和91.55%,能够满足后续分析要求。

表1 测序数据统计
2.2 差异表达基因分析
根据FDR<0.05且Fold change>3的标准,以健康桑树叶片为参照,筛选统计两样本间的差异表达基因(different expression genes,DEGs),结果1 394个基因具有差异表达,其中1 032个表达上调,362个表达下调(图1),575个基因在MO样本中有表达,1 250个在MV样本中具有表达,两样本中均有表达基因数431个(图1)。
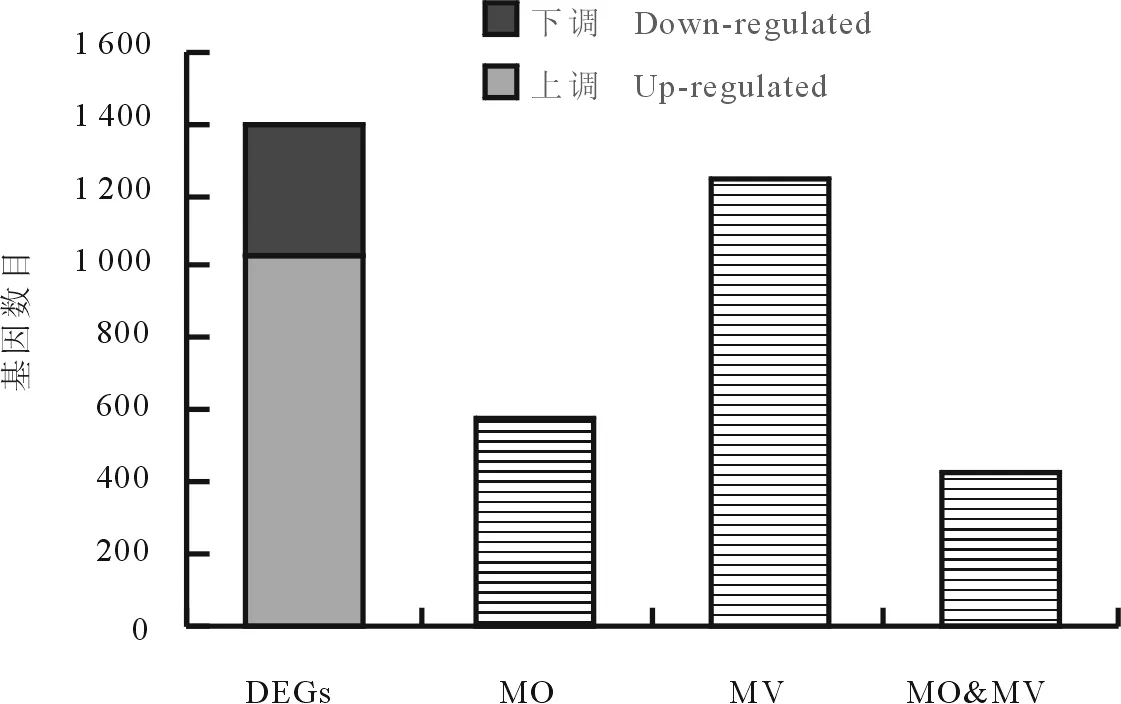
图1 两样本基因表达差异统计(FDR<0.05)
2.3 差异表达基因的GO功能分类
进一步分析两样品DEGs的GO功能分类分析,结果显示(图2),分子功能(molecular function)分类中,主要聚集在结合(binding)和催化活性(catalytic activity);细胞组分(cell component)分类主要聚集在细胞(cell)和膜组分(membrane part);生物学过程(biological process)分类中主要集中在细胞进程(cellular process)和代谢过程(metabolic process)。

图2 差异表达基因的GO注释
2.4 差异表达基因的KEGG代谢通路分析
以KEGG数据库作参考进行分析,结果其中的399个DEGs参与了代谢通路的6大类39个亚类175小类。图3A为KEGG大类分区6大类分别占比例,可以看到,6大类中,代谢类通路所占比例最大,为43.30%,其次是人类疾病和遗传信息处理相关通路,占比分别为16.29%和13.93%,细胞过程、生物系统和环境信息处理相关的通路,占比为8.25%、8.79%和9.43%。
图3B是小类分区中存在大于10个DEGs的代谢通路的情况,其中小类中数量最多的是KO03010,来自遗传信息处理大类的翻译亚类的核糖体,总数达76个。KO00010KO00380来自代谢类通路,KO00010、KO00620、KO00640和KO00650为碳水化合物代谢亚类,KO00190、KO00680和KO00910来自能量代谢亚类,KO00280和KO00380来自氨基酸代谢亚类,这3个亚类分别占代谢通路的10.61%、7.07%和8.36%,是代谢类通路的主要组成部分。另外来自环境信息处理大类信号转导亚类的KO02020数量也较多,为32个。
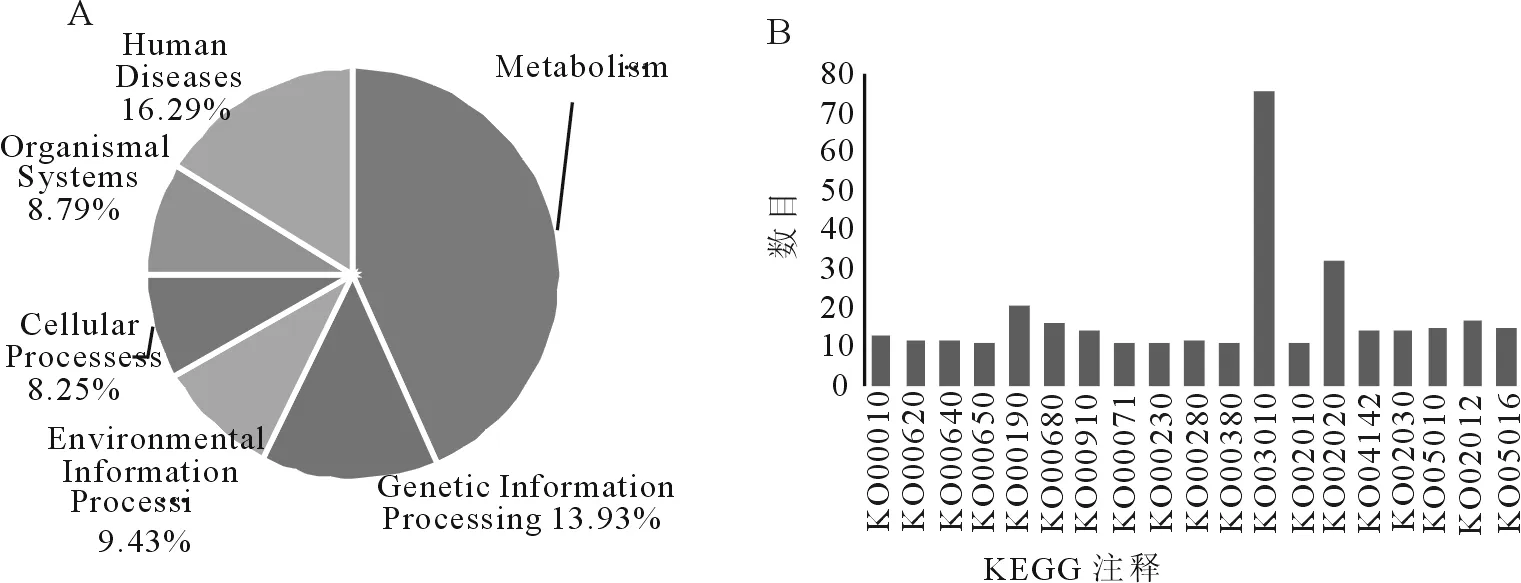
图3 桑树转录组DEGs的KEGG代谢途径分类
2.5 抗病相关DEGs基因分析
植物抗病基因(R基因)起源于植物和动物共有的一种古老的免疫系统,在识别病原体特异性蛋白质方面起着关键作用。进一步依据基因的功能注释和已有的基因功能的报道,对可能与植物病害相关基因进行筛选,结果获得89个基因(表2,续表2),这些基因主要涉及丝氨酸/苏氨酸蛋白激酶(Serine/theronine-protein kinase)、抗病蛋白(Putative disease resistance protein)、受体类蛋白激酶(receptor-like protein kinase)、脂质氧化酶等37类。以正常未患病桑叶样品为对照,结果显示:抗病相关DEGs中,50个基因上调表达,39个基因下调表达。

表2 抗病相关差异表达基因数量统计

续表2 抗病相关差异表达基因数量统计
3 讨论
桑树基因组测序的完成为系统、深入地挖掘桑树功能基因,揭示对病原应答的分子机制提供了重要基础条件[17]。在有参考基因组的基础上进行转录组测序和数据分析,可以获得更精准的序列数据信息。川桑基因组序列共注释基因29 338个[18],笔者研究测序获得了健康和MYDD桑树叶片两个样本的11.57Gb数据,共得到62 953个UniGenes,注释了27 464个基因,覆盖了川桑基因组注释的83%的基因,进一步去除植物的组织特异表达,说明测序数据和组装效果好,注释信息丰富。将27 464个基因与COG数据库识别并对直系同源基因产物进行分类,结果有21 844条与数据库中的基因具有相似性,分别被注释到23个COG功能分类,涉及了桑树大部分的生命活动,与10~20%谱系特有基因结果一致[19]。同时发现了378个在川桑中没有注释的基因,而通过巴旦木、葡萄、可可、野草莓等植物的同源序列进行了注释。这说明桑属植物内具有较高的遗传多样性,也为桑属植物多样性的研究和优良育种基因的鉴定提供了遗传背景参考。
为明确植原体侵染对桑树叶片转录组的影响,本研究筛选到了一批在健康和MYDD桑树叶片中存在差异表达的UniGenes,找到了2 766条和3 570条分别在健康和MYDD桑树叶片中单独表达的UniGenes。结合UniGenes的GO功能差异富集,找到了富集量超过50条UniGenes的41个GO分类条目,7 909条对应关系,共涉及到11个亚类,发现植原体侵染相关的差异UniGenes参与了分子功能的结合类在其中占比最多,其次是生物学过程的细胞进程类和细胞组分的细胞等生物学过程,这与植原体侵染导致植物内源激素失衡的研究有一定的对应关系。植原体能够造成寄主的内源激素紊乱,细胞分裂素含量增加,赤霉素含量降低,玉米素/生长素比值增加,而内源激素在调控植物细胞的分裂、伸长、分化、生长等生物学过程中起着重要作用[20]。
进一步对健康和MYDD病植株的差异表达UniGenes基因与生命和环境联系起来,进行基于KEGG的基因和基因组的代谢通路富集分析,找到了8个差异显著的通路,涉及40余个蛋白。由于基因是相互影响的,1个基因往往有多个功能,而具体功能往往是多个基因共同作用的结果[21],所以富集分析的结果总是会涉及特别多的通路。而且植原体的侵染可以对寄主植物产生多方面的影响,如蛋白质和氨基酸组成、IAA信号转导、过氧化物酶、酯酶等酶系统,次生代谢的生物合成[22]。本研究的KO00910和KO00250途径的GAD、GdhA等谷氨酸代谢相关基因与在甜樱桃、枣中得到研究结果一致[23~24],KO00908玉米素生物合成Zeatinbiosynthesi也在多个物种中得到一致的结果[24~25]。
Cardiac muscle contraction(KO04260)是一个在哺乳动物中注释的细胞通路,这部分基因在植物中多是叶绿体和线粒体基因。叶绿体是植物光合作用的重要器官,在植物的生长发育中具有重要作用,许多植物在被植原体感染后,光合作用显著降低,碳水化合物的积累被抑制[26]。本研究发现部分参与光合作用的基因在MYDD桑树叶片中的表达出现下调,这可能造成电子传递受阻,引起感染植物的光合作用的降低[27]。
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
作文大王·低年级(2022年4期)2022-04-23
中国典型病例大全(2022年7期)2022-04-22
科学导报(2021年29期)2021-06-03
初中生世界·七年级(2020年8期)2020-09-06
中国瓜菜(2019年8期)2019-09-19
科海故事博览·下旬刊(2019年6期)2019-04-16
湖北农业科学(2017年4期)2017-03-28
江苏农业科学(2016年11期)2017-03-21
江苏农业科学(2017年1期)2017-02-27
