
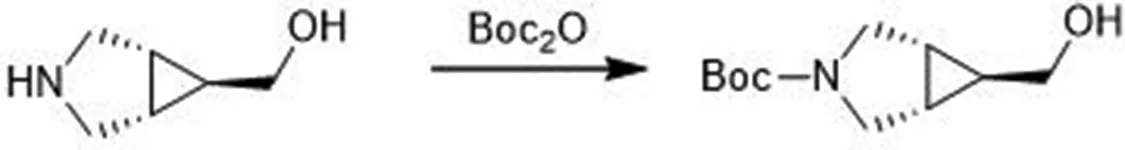
(1R,5S,6r)-6-甲酰基-3-氮杂双环[3.1.0]己烷-3-羧酸叔丁酯的合成
2022-11-23 08:01杨葵华何冀川
绵阳师范学院学报 2022年11期
杨葵华,何冀川,魏 娜
(绵阳师范学院化学与化学工程学院,四川绵阳 621000)
0 引言
氮杂环类化合物以独特的生物活性、低毒性和高内吸性闻名,是医药、农药和含能材料等许多领域中不容缺少的有机中间体和化工原料[1].独特小分子药物中约59%都含有氮杂环化合物,氮杂环化合物在多肽中的应用也极为广泛.目前,将一些构型受限的氨基酸用到多肽的设计与合成中,已经取得了较大成功[2-3].含氮化合物也较易作为结构的修饰物,很方便地引入各种基团,使它具有不同的性能[4].
(1R,5S,6r)-6-甲酰基-3-氮杂双环[3.1.0]己烷-3-羧酸叔丁酯是一种含氮杂环化合物,是用于合成多种生物活性药物的医药中间体之一.目前关于本文所述中间体的合成和用途还缺少综合性的报道,其在新药开发中的相关应用尚在研究和试验期间.
1 实验原理
合成路线一原理:醋酸铑催化N-Boc-3-吡咯啉与重氮乙酸乙酯发生环丙烷化反应[5]合成氮杂双环,经硼氢化锂还原为醇,再由DMP(Dess-Martin高碘烷)氧化成醛[6].

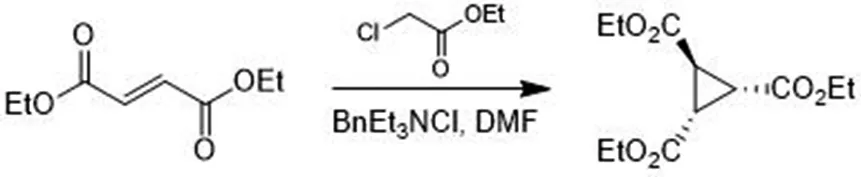
合成路线二原理:氯乙酸乙酯和富马酸二乙酯加成反应后[7],在苄基三乙基氯化铵的催化下[8]拔掉氯乙酸乙酯中靠近氯原子的碳上的一个氢原子,形成碳负离子,再由富马酸二乙酯进攻碳负离子制备1,2,3-三甲酸乙酯环丙烷.在碱性条件下酯基水解,然后用盐酸酸化成三元酸,将三元酸与醋酸酐反应,再与苄胺闭环后酯化,用红铝将酯选择性还原为相应醇[9],用湿钯碳加氢脱苄基[10]后与二碳酸二叔丁进行氨基保护反应[11],再用DMP氧化为目标醛.
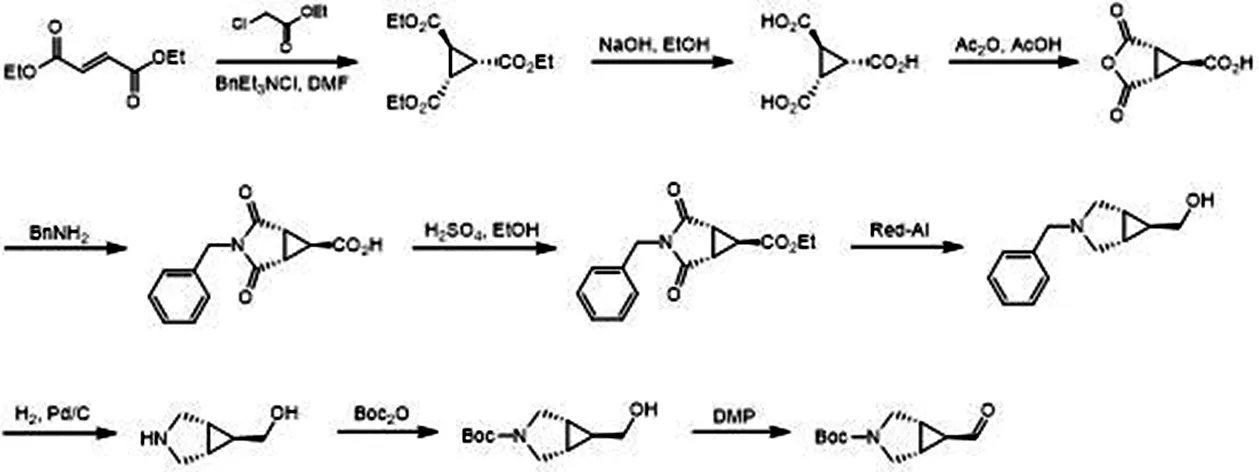
2 实验部分
2.1 仪器与试剂
所用仪器有:NMR Magnet System恒温磁力搅拌器,暗箱式紫外分析仪,温度控制仪,快速制备色谱仪,电子分析天平,薄层层析硅胶板,旋转蒸发器,布鲁克AVANCE NEO核磁共振(NMR)波谱仪.
所用试剂均为分析纯:N-Boc-3-吡咯啉,硼氢化锂,双(2-甲氧基乙氧基)氢化铝钠70%甲苯溶液,四水合L(+)-酒石酸钾钠,苄基三乙基氯化铵,富马酸,二乙酯,重氮乙酸乙酯,氯乙酸乙酯,戴斯马丁试剂,二碳酸二叔丁酯,二聚醋酸铑,三乙胺苄胺,10%湿Pd/C,无水碳酸钾,氢氧化钠碳酸氢钠,浓硫酸,乙酸酐,盐酸,乙酸,丙酮,无水甲基叔丁基醚,无水二氧六环,无水三氯甲烷,无水四氢呋喃,无水二氯甲烷,无水石油醚,乙酸乙酯,无水甲苯,无水甲醇,无水乙醇,无水DMF.
2.2 合成线路一
2.2.1 (1R,5S,6R)-3-叔丁氧羰基-6-甲酸乙酯-3-氮杂双环[3.1.0]己烷的合成
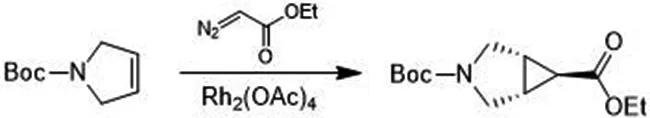
称量17.00 gN-Boc-3-吡咯啉和0.532 8 g醋酸铑于500 mL茄形瓶中,加入85.0 mL1,2-二氯乙烷溶解.称量18.00 g重氮乙酸乙酯于烧杯中,加入85.0 mL1,2-二氯乙烷溶解,转入250 mL恒压滴液漏斗中,在80 ℃于氮气保护下均匀滴加茄形瓶中,耗时约72 h.点硅胶板监测[12],展开剂为石油醚与乙酸乙酯,体积比为5∶1,碘熏产物均无紫外.硅胶板结果显示N-Boc-3-吡咯啉完全反应且有三个极性更大的产物生成,其中极性最小的产物有紫外.过滤除去醋酸铑,旋转蒸干,得30.00 g粗产品(Rf=0.3).
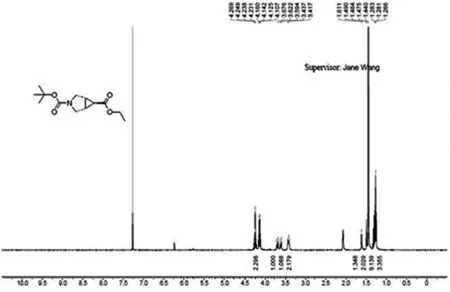
图1 (1R,5S,6R)-3-叔丁氧羰基-6-甲酸乙酯-3-氮杂双环[3.1.0]己烷1H核磁共振氢谱图Fig.1 1H NMR spectrum of (1R, 5S, 6R) - 3-tert-butoxycarbonyl-6-carboxylic acid ethyl ester-3-azabicyclo [3.1.0] hexane
将上述30.00 g粗产品加适量乙酸乙酯溶解后,再加入约60 g硅胶装柱待用.分别用100 mL乙酸乙酯、150 mL石油醚润洗过柱机,将220 g标准硅胶柱用石油醚润湿至体系无气泡,上样.乙酸乙酯从0%缓慢升至8%,全收集.将柱色谱中有紫外吸收的峰完全流出以后所收集的各试管溶液每隔五支点样,碘熏,将碘熏能观察到的点再点样,与N-Boc-3-吡咯啉比对,选中目标点(Rf=0.3),将目标点前后四管点样碘熏观察,收集有目标点的所有样液,旋转蒸干,得5.010 0 g无色透明油状物.核磁共振氢谱[13]见图1,1HNMR(400 MHz,CDCl3)∶δ4.12(q,J=7.2 Hz,2H),3.58-3.69(m,2H),3.40(m,2H),2.05(m,2H),1.46(t,J=3.2 Hz,1H),1.42(s,9H),1.25(t,J=7.2 Hz,3H).
2.2.2 (1R,5S,6R)-3-叔丁氧羰基-6-甲醇-3-氮杂双环[3.1.0]己烷的合成

称量0.700 0 g 2.2.1第一步产物于50 mL茄形瓶中,加入15 mL无水THF溶解,将温度降到0 ℃后缓慢滴加
0.196 0 gLiBH4.全部溶解后升温至50 ℃,缓慢滴加2 mL无水甲醇,50 ℃条件下反应0.5 h.点硅胶板监测,展开剂为石油醚与乙酸乙酯,体积之比为3∶1,碘熏.硅胶板显示原料少许没反应完,能观察到一个比反应物极性更大的主点(Rf=0.1).
将反应液缓慢滴加进10.0 mLH2O中,再加入适量水至体系无气泡产生,有白色固体产生,溶液变浑浊.将混合液用旋转蒸发仪将有机相旋干,向残留水相中滴加6 mL 的1 mol·L-1盐酸溶液至白色固体全部溶解,加入80.0 mLH2O,用20 mL乙酸乙酯萃取四次.将有机相用饱和食盐水洗一次,无水硫酸钠干燥静置,将有机相用旋转蒸发仪旋干,得0.360 0 g粗产品.

图2 (1R,5S,6R)-3-叔丁氧羰基-6-甲醇-3-氮杂双环[3.1.0]己烷1H核磁共振氢谱图Fig.2 1H NMR spectrum of (1R, 5S, 6R) - 3-tert-butoxycarbonyl-6-methanol-3-azabicyclo [3.1.0] hexane
将4 g标准硅胶柱用石油醚润湿至体系无气泡,将0.360 0 g粗产品加适量乙酸乙酯溶解,加入约1 g硅胶粉拌样装柱,然后用乙酸乙酯浓度从0%缓慢升至30%,全收集.得到两个产品,分开收集用旋转蒸干,极性更大的产物LCMS显示:在波长为220 nm,保留时间为0.686 min时产生峰,并且该峰的m/z值正好为目标产物脱掉叔丁基的数值,纯度达到100%.即极性更大的为目标产物(Rf=0.1),得0.240 3 g,理论产量584.75 mg,产率为41.10%.核磁共振氢谱见图2,1HNMR(400 MHz,CDCl3)∶δ3.61-3.54(m,3H),3.52-3.50(m,1H),3.47-3.33(m,2H),1.47-1.41(m,11H),0.94(m,1H).
2.2.3 (1R,5S,6r)-6-甲酰基-3-氮杂双环[3.1.0]己烷-3-羧酸叔丁酯的合成

在20 ℃条件下,将0.240 3 g上步产品溶于装有12.0 mLCH2Cl2的50 mL茄型瓶中,加入0.715 9 g戴斯马丁试剂,恒温反应2 h.点硅胶板监测,硅胶板显示原料反应完全,能观察到一个比反应物极性更小的主点(Rf=0.5).
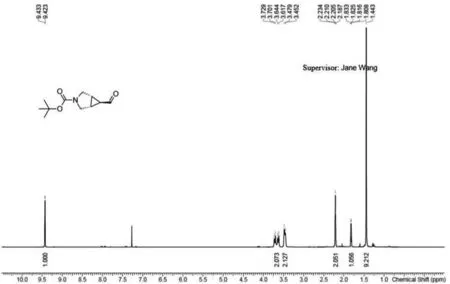
图3 (1R,5S,6r)-6-甲酰基-3-氮杂双环[3.1.0]己烷-3-羧酸叔丁酯1H核磁共振氢谱图Fig.3 1H NMR spectrum of (1R, 5S, 6R)-6-formyl-3-azabicyclo[3.1.0] hexane-3-carboxylic acid tert butyl ester
将反应液过滤,乙酸乙酯冲洗,滤液旋转蒸干得0.220 0 g粗产品,将粗产品用1,2-二氯乙烷溶解,硅胶粉拌样上样.乙酸乙酯浓度从0%缓慢升至30%,全收集.将目标点收集旋转蒸干,得0.111 0 g无色油状产品,静置一夜后变为白色固体,理论产量286.67 mg,产率为38.70%.LCMS显示:在波长为220 nm,保留时间为0.715 min时产生峰,峰值为目标产物脱掉叔丁基的值,纯度为100%.核磁共振氢谱见图3,1HNMR(400 MHz,CDCl3)∶δ9.43(d,J=4 Hz,1H),3.72(d,J=11 Hz,1H),3.64(d,J=11 Hz,1H),3.47-3.45(m,2H),2.23-2.18(m,2H),1.82(q,J=4 Hz,1H),1.44(s,9H).
2.3 合成线路二
2.3.1 1,2,3-三甲酸乙酯环丙烷的合成
称量88.30 g无水碳酸钾,0.661 5 g苄基三乙基氯化铵,量取250 mL无水DMF于1 L三口烧瓶中,加热至40 ℃.称量50.00 g富马酸二乙酯,48.04 g氯乙酸乙酯,50 mL无水DMF做溶剂装于250 mL恒压滴液漏斗,逐滴加入约8 h,在40 ℃条件下反应15 h,体系由无色变为浅黄再变为深黄.
将反应液倒入2.5 L烧杯中,加入约1 L水搅拌,每次用500 mL无水甲基叔丁基醚萃取三次.将有机相用水洗两次,饱和食盐水洗一次,无水硫酸钠干燥,静置旋转蒸干,得74.90 g黄色油状物粗产品.将74.90 g粗产品减压蒸馏纯化,收集120~130 ℃蒸馏出来的组分,得57.97 g无色油状液体,产率为77.29%.产物核磁共振氢谱见图4,1HNMR(400 MHz,CDCl3)∶δ4.23-4.11(m,6H),2.76(t,J=5.4 Hz,1H),2.54(d,J=5.4 Hz,2H),1.29(dt,J=14.3,7.5 Hz,9H).
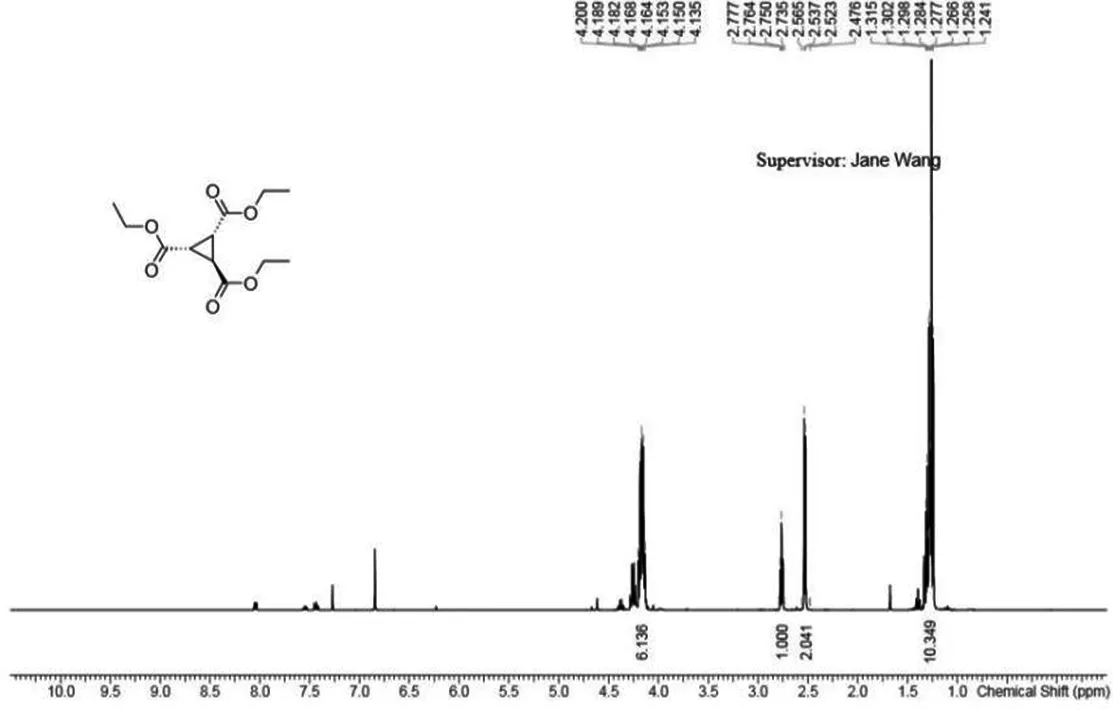
图4 1,2,3-三甲酸乙酯环丙烷1H核磁共振氢谱图Fig.4 1H NMR spectrum of 1,2,3-ethyl tricarboxylate cyclopropane
2.3.2 1,2,3-三甲酸环丙烷的合成
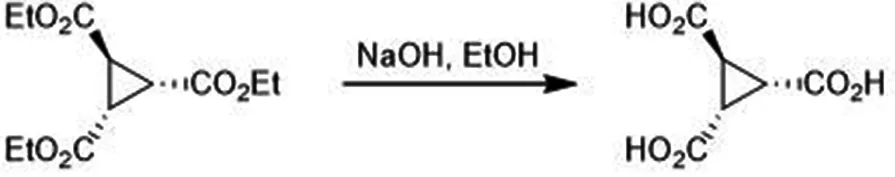
将30.0 mL无水乙醇和61.7 mL10 mol·L-1的NaOH溶液于100 mL茄型瓶中混合,加热至50 ℃,将57.97 g产物逐渐滴加入,保持温度低于回流温度.滴加完成后升温至90 ℃,回流2 h后体系成浆状,再用12 moL·L-1的盐酸将溶液pH调至1,冷却至室温旋转蒸干,得80.00 g粗产品.将80.00 g粗产品加480.0 mL丙酮溶解,升温至60 ℃回流打浆2 h,趁热过滤,旋转蒸干得10.90 g产品,理论产量39.07 g,产率为27.90%.核磁共振氢谱:1HNMR(400 MHz,DMSO)∶δ12.77(s,3H),2.41-2.29(m,3H).
2.3.3 (1R,5S,6S)-2,4-二氧代-6-甲酸-3-氧杂二环[3.1.0]己烷的合成
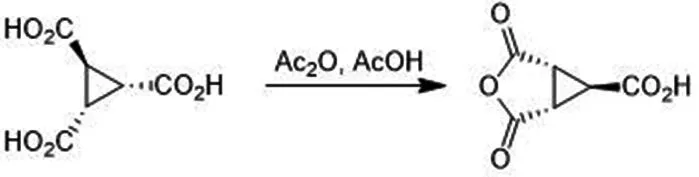
将第二步的10.90 g三元酸产品加入23.7 mL醋酸和8 mL醋酸酐溶解,升温至120 ℃,回流搅拌2 h.旋转蒸干醋酸和醋酸酐,加少量甲苯将体系旋至无水状态,再加少量甲苯在0 ℃下打浆30 min,过滤,用甲苯洗涤滤饼.滤饼旋转蒸干得8.84 g产品,产率90.39%.核磁共振氢谱见图5,1HNMR(400 MHz,DMSO-d6)∶δ13.4(s,1H),3.24(d,J=3.2 Hz,2H),3.07(d,J=3.2 Hz,1H).
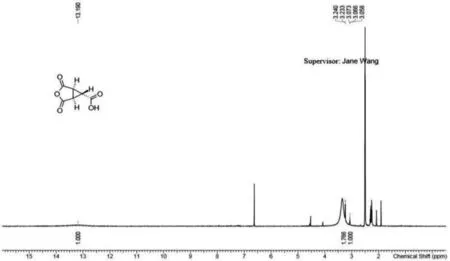
图5 (1R,5S,6S)-2,4-二氧代-6-甲酸-3-氧杂二环[3.1.0]己烷1H核磁共振氢谱图Fig.5 1H NMR spectrum of (1R, 5S, 6S) - 2,4-dioxo-6-carboxylic acid-3-oxabicyclo [3.1.0] hexane
2.3.4 (1R,5S,6R)-3-苄基-6-甲酸-2,4-二氧代-3-氮杂双环[3.1.0]己烷的合成
将8.84 g第三步产品溶于48.0 mL丙酮中,加入8 mL三乙胺和7 mL苄胺,室温反应3 h后用旋转蒸发干,再加入2.79 g醋酸钠和25.5 mL醋酸酐,120 ℃条件下回流1 h,冷至室温后旋转蒸干,加入适量水将反应体系淬灭,用1 moL·L-1盐酸将体系pH调至2,有棕褐色固体析出,将固体旋干,得6.90 g粗产品.磁共振氢谱见图6,1HNMR(400 MHz,DMSO-d6)∶δ13.17(s,1H),7.07-7.42(m,5H),4.38(s,2H),2.95(s,2H),2.64(s,1H).

图6 (1R,5S,6R)-3-苄基-6-甲酸-2,4-二氧代-3-氮杂双环[3.1.0]己烷1H核磁共振氢谱图Fig.6 1H NMR spectrum of (1R, 5S, 6R)-3-benzyl-6-carboxylic acid-2,4-dioxo-3-azabicyclo [3.1.0] hexane
2.3.5 (1R,5S,6R)-3-苄基-6-甲酸乙酯-2,4-二氧代-3-氮杂双环[3.1.0]己烷的合成

将6.90 g第四步粗产品与68.0 mL乙醇混合,升温至60 ℃后滴加600 μL浓硫酸,并于60 ℃反应6 h.将反应液降至0 ℃有棕褐色固体析出,过滤,得7.00 g粗产品.LCMS显示:在波长为220 nm,保留时间为0.774 min时出现主峰,峰值为目标产物与乙醇的总和峰值,纯度为79.72%.还有4.64%为原料未反应完全,以及少许其他杂质.
将80 g标准硅胶柱用石油醚润湿至体系无气泡,将7.00 g粗产品乙酸乙酯溶解,加入约20 g硅胶装柱,然后将乙酸乙酯浓度从0%缓慢升至30%冲洗,全收集.目标点用旋转蒸干,得5.00 g白色产品,产率为65.02%.核磁共振氢谱:1HNMR(400 MHz,CDCl3)∶δ 7.40-7.23(m,5H),4.51(s,2H),4.18(q,J=7.1 Hz,2H),2.87(d,J=2.7 Hz,2H),2.28(t,J=2.7 Hz,1H),1.27(t,J=7.1 Hz,3H).
2.3.6 (1R,5S,6R)-3-苄基-6-甲醇-3-氮杂双环[3.1.0]己烷的合成
将5.00 g产物溶于35.0 mL无水DMF中,加入30.5 mL双(2-甲氧基乙氧基)氢化铝钠70%甲苯溶液,20 ℃条件下反应16 h.点硅胶板监测,用石油醚:乙酸乙酯体积比为5∶1展开,茚三酮烘烤.硅胶板显示原料反应完全,能观察到两个比反应物极性更大的主点(Rf=0.4).
向反应液中缓慢滴加过量30%酒石酸钾钠,74.4 mL酒石酸钾钠溶液,将混合物搅拌30 min,静置分离两相.用20 mL乙酸乙酯将水相萃取三次.将有机相用饱和食盐水洗一次,无水硫酸钠干燥静置,旋转蒸干,得3.87 g棕色油状粗产品.核磁共振氢谱见图7,1HNMR(400 MHz,CDCl3)∶δ 7.50-7.09(m,5H),3.52(s,2H),3.38(d,J=7.3 Hz,2H),2.96(d,J=8.8 Hz,2H),2.54(s,1H),2.35(d,J=8.6 Hz,2H),1.60-1.50(m,1H),1.22(s,2H).LCMS显示:在波长为220 nm的整个分离时间内,不断出峰,且峰值所对应产物无法判断,只有在保留时间为0.660 min时出现明显的目标产物与水的总和值的峰,纯度为4.32%.
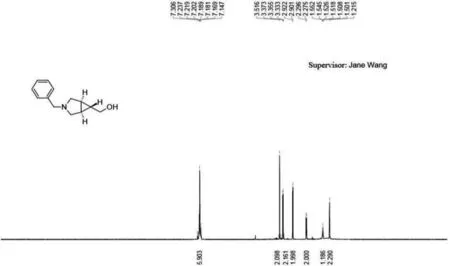
图7 (1R,5S,6R)-3-苄基-6-甲醇-3-氮杂双环[3.1.0]己烷1H核磁共振氢谱图Fig.7 1H NMR spectrum of (1R, 5S, 6R)-3-benzyl-6-methanol-3-azabicyclo [3.1.0] hexan
2.3.7 (1R,5S,6R)-6-甲醇-3-氮杂双环[3.1.0]己烷的合成

将3.87 g第六步产物溶于80.0 mL无水甲醇中,在氮气保护下加入0.387 0 g10%湿钯碳,向体系通入氢气置换氮气,使整个体系于氢气状态下反应16 h.点硅胶板监测,展开剂为石油醚∶乙酸乙酯,体积比为4∶1,茚三酮烘烤.硅胶板显示原料反应完全,有新点产生.
反应液用硅藻土过滤掉钯碳,将滤液旋转蒸干,得1.610 0 g白色固体,理论产量3.03 g,产率为53.13%.核磁共振氢谱:1HNMR(400 MHz,CDCl3)∶δ 3.52-3.50(d,J=7 Hz,2H),2.99(d,J=11 Hz,2H),2.91-2.86(bd,J=12 Hz,2H),1.35(m,2H),0.89(m,1H),有少量杂质.
2.3.8 (1R,5S,6R)-3-叔丁氧羰基-6-甲醇-3-氮杂双环[3.1.0]己烷的合成
将1.610 0 g第七步产物溶于15 mL无水二氧六环和15 mL饱和碳酸氢钠溶液中,加入7 mL二碳酸二叔丁酯溶液,20 ℃条件下反应16 h.将反应液点硅胶板监测,展开剂为石油醚为乙酸乙酯,体积比为1∶1,茚三酮烘烤.硅胶板显示有一个新主点产生(Rf=0.3).
向反应液中加150 mL水,用40 mL乙酸乙酯萃取四次.将有机相用饱和食盐水洗一次,无水硫酸钠干燥,旋转蒸干得6.50 g粗产品.将40 g标准硅胶柱用石油醚润湿至体系无气泡,将6.50 g粗产品加适量1,2-二氯乙烷溶解加入约2 g硅胶装柱上样,然后用乙酸乙酯浓度从0%缓慢升至50%冲洗,全收集.将主点旋转蒸干,得2.070 0 g产品,产率为68.32%.核磁共振氢谱见图8,1HNMR(400 MHz,CDCl3)∶δ 3.61-3.54(m,3H),3.52-3.50(m,1H),3.47-3.33(m,2H),1.47-1.41(m,11H),0.94(m,1H).LCMS显示:在波长为220 nm,保留时间为0.675 min和0.808 min时产生主峰,并且峰值正好为目标产物脱掉叔丁基的值,纯度达到97.20%.
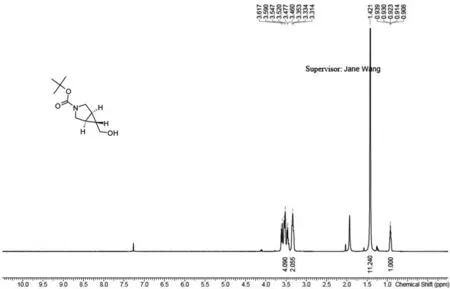
图8 (1R,5S,6R)-3-叔丁氧羰基-6-甲醇-3-氮杂双环[3.1.0]己烷1H核磁共振氢谱图Fig.8 1H NMR spectrum of (1R, 5S, 6R)-3-tert-butoxycarbonyl-6-methanol-3-azabicyclo [3.1.0] hexane
2.3.9 (1R,5S,6r)-6-甲酰基-3-氮杂双环[3.1.0]己烷-3-羧酸叔丁酯的合成

将2.070 0 g第八步产品溶于90.0 mL二氯甲烷于250 mL三口烧瓶中,在20 ℃加入6.17 g戴斯马丁试剂,反应2 h.点硅胶板监测,展开剂为石油醚与乙酸乙酯,体积比为3∶1,碘熏.硅胶板显示反应完全,能观察到一个比反应物极性更小的主点产生(Rf=0.5).
将12 g标准硅胶柱用石油醚润湿至体系无气泡,将乙酸乙酯冲洗过的滤液旋转蒸干得粗产品,将粗产品用1,2-二氯乙烷溶解硅胶粉拌样上样,然后用乙酸乙酯浓度从0%缓慢升至30%冲洗全收集,用50 mL茄形瓶在旋转蒸发仪上旋干,得1.360 0 g无色油状产品,产率为52.71%.核磁共振氢谱见图9,1HNMR(400 MHz,CDCl3)∶δ 9.43(d,J=4 Hz, 1H),3.72(d,J=11 Hz,1H),3.64(d,J=11 Hz,1H),3.47-3.45(m,2H),2.23-2.18(m,2H),1.82(q,J=4 Hz,1H),1.44(s,9H).

图9 (1R,5S,6r)-6-甲酰基-3-氮杂双环[3.1.0]己烷-3-羧酸叔丁酯1H核磁共振氢谱图Fig.9 1H NMR spectrum of (1R, 5S, 6R)-6-formyl-3-azabicyclo [3.1.0] hexane-3-carboxylic acid tert butyl ester
3 结果与讨论
合成结果对比见表1.路线一步骤简单但存在较多不足,重氮乙酸乙酯是一种不稳定的物质,加热易引起分解和爆炸,制约了工业化生产;其次还原过程中用了较危险且具有腐蚀性,遇水反应迅速易制爆的硼氢化锂试剂;最后合成过程中使用了较昂贵的醋酸铑试剂,一克即需五百余元,并且反应时间久,产率低,大大增加生产成本.路线二虽然步骤较多,反应较繁琐,合成时间也比较长,但富马酸二乙酯和氯乙酸乙酯价格便宜,而且总收率较高.

综上,通过比较原料成本,危险性,步骤,总耗时与总收率可得:路线一不适合用于工业生产,路线二是更加经济可行的路线,符合大规模工业生产要求.
猜你喜欢
科学导报(2022年41期)2022-07-13
安徽农学通报(2022年8期)2022-05-06
金沙江文艺(2022年1期)2022-02-04
化学教与学(2021年12期)2021-02-18
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24
时代邮刊·下半月(2019年8期)2019-09-10
热带农业工程(2017年2期)2017-08-29
吉林农业(2017年5期)2017-05-13
家用汽车(2016年12期)2017-02-09
江苏农业科学(2015年1期)2015-04-17
