
限制性内切酶的无细胞快速制备研究
2023-09-16 03:06刘晚秋季向阳许慧玲卢屹聪李健
合成生物学 2023年4期
刘晚秋,季向阳,许慧玲,卢屹聪,李健
(上海科技大学物质科学与技术学院,上海 201210)
20 世纪50 年代,研究人员发现在细菌中存在一套可以切割噬菌体核酸序列,同时能保护自身核酸序列不被切割,从而抵御外源侵染的机制,这套机制包括作用于DNA 特定序列的内切酶和识别自身基因组上相同序列而进行甲基化修饰保护的甲基化酶,研究人员将这一机制命名为限制性修饰系统(restriction modification system,RM system),据此将这一系统中核酸内切酶命名为限制性内切酶(restriction endonuclease)[1‑2]。随后,研究人员Werner Arber、Daniel Nathans和Hamilton O.Smith深入研究了这类酶的催化机制,发现其能识别特定的核苷酸序列,并对每条链中特定位点的两个脱氧核糖核苷酸之间的磷酸二酯键进行切割,这一发现获得了1978 年的诺贝尔生理学和医学奖[3‑5]。在此后的研究中,研究人员发现这类酶在自然界中广泛存在,并根据酶的功能特性、大小及反应时所需要的辅因子等将它们分为ClassⅠ~Ⅳ四大类。ClassⅡ类限制性内切酶是4类酶中唯一不需要ATP、仅需Mg2+就能实现切割功能的酶,其识别序列多为较短的回文序列,剪切位点通常即为识别序列[6‑8],广泛应用于基因/染色体的结构鉴定和图谱绘制、基因的遗传操作以及核酸蛋白相互作用等研究,是生物学研究中必不可少的工具之一[8‑12]。
最早期限制性内切酶的制备是通过产生菌大量发酵后经过多步生物化学方法分离纯化获得目标酶[4],步骤烦琐、技术要求高、产量低且酶活稳定性较差;目前限制性内切酶主要通过异源重组表达纯化获得[13‑14]。但在异源表达过程中,限制性内切酶对特定DNA 序列的切割特性严重抑制了宿主的正常生长和重组蛋白的表达。甲基化保护重组表达是限制性内切酶制备的主要策略,即共表达甲基化酶或构建甲基化酶表达宿主后再表达目标酶,以甲基化酶修饰宿主DNA 中的酶切位点,从而保证宿主的正常生长和目标蛋白的稳定表达[15‑17]。由于底物识别特异性高,每个限制性内切酶都有与之相对应的甲基化酶,这使得每一个限制性内切酶的制备都需要构建相应的甲基化酶共表达体系。另一方面,大多数具有类似催化功能的酶蛋白在氨基酸序列或结构上具有一定的同源性而形成酶蛋白家族,据此可以进行新成员的挖掘、功能演化等研究,而限制性内切酶没有序列和结构同源的酶蛋白家族,即使具有相似DNA切割活性的限制性内切酶,其进化轨迹也相对独立[18]。这样“一酶一策略”的原则严重限制了此类酶的制备效率及新型限制性内切酶的挖掘和应用。因此,对限制性内切酶的研究亟需一个简单、普适性高的方法。
近年来,无细胞蛋白合成(cell‑free protein synthesis, CFPS)技术快速发展,成为蛋白表达的新平台,也为合成生物学和生物合成研究提供了新手段[19‑22]。无细胞蛋白合成的基本原理是将细胞生长与蛋白合成分开进行,首先以最佳生长状态的细胞(如微生物)制备出高活性的细胞提取物(包含与蛋白合成相关的核糖体、辅助因子等活性成分),再向其中添加能量(ATP)再生系统、基因模板(如DNA 或mRNA)以及底物和其他辅助因子等,即可快速合成目标蛋白。与传统体内蛋白表达系统相比,CFPS 体系具有反应条件可控、反应速率快、蛋白产量高、产物易分离、无细胞毒害作用等诸多优点。鉴于以上优势,近年来CFPS 体系已被成功应用于合成药物蛋白、膜蛋白、金属蛋白和在蛋白中定点引入非天然氨基酸等,显示了其强大和广泛的应用前景[23‑29]。
本研究将CFPS 技术应用于限制性内切酶的表达制备。CFPS 技术将宿主生长和蛋白表达时空分离,因而限制性内切酶的无细胞表达过程中不存在细胞毒性的影响,不需要甲基化酶的共表达,同时以线性DNA为表达模板也避免了表达载体构建过程中泄露表达对克隆宿主的细胞毒性,将大大简化限制性内切酶的重组表达。本研究中选择限制性内切酶EcoRⅠ[30]、BamHⅠ[31]和BsaⅠ[32]为研究对象。EcoRⅠ和BamHⅠ分别来自于Escherichia coliRY13和Bacillus amyloliquefaciensH,是典型的ClassⅡ类限制性内切酶,也是分子生物学中使用频率最高的几个限制性内切酶。BsaⅠ来自于Bacillus stearothermophilus6‑55,其切割位点位于识别位点之后,切割后产生黏性末端,属于ClassⅡTypeⅡs限制性内切酶,是Golden Gate技术进行DNA 组装的必需成员[33],受限于其制备、研发技术和成本,ClassⅡTypeⅡs 类限制性内切酶的价格高于其他ClassⅡ类限制性内切酶。EcoRⅠ、BamHⅠ和BsaⅠ的无细胞表达可以验证CFPS 技术在不同限制性内切酶制备研究中的适用性。为此,我们以大肠杆菌无细胞提取物为基础,以线性DNA 片段为表达模板,经过一系列的调控和优化,成功构建了限制性内切酶的通用型体外表达及制备体系(图1),实现了ClassⅡ类3 个限制性内切酶的体外简单快速制备。无细胞制备限制性内切酶的方法突破了传统技术中“一酶一策略”的表达原则,理论上可以适用于所有限制性内切酶的表达与制备,将为今后限制性内切酶的高效合成、快速发掘、筛选进化等深入研究和广泛应用提供崭新的思路。
1 材料与方法
1.1 材料
1.1.1 菌株、质粒和引物
E. coliDH5α 用于常规的克隆和质粒扩增,E. coliBL21 Star(DE3)用于细胞提取物制备。质粒pJL1、pET28a和pSUMO用于无细胞表达线性模板构建,pG‑KJE8(Takara)用于促进蛋白可溶表达。限制性内切酶基因EcoRⅠ(GenBank:AAA26371.1)、BamHⅠ(GenBank:QDF53662)和BsaⅠ(GenBank:AAR96018.1)由苏州金唯智生物科技有限公司合成并构建至质粒pUC‑GW‑Kan 上。本研究所用寡核苷酸引物[由生工生物工程(上海)股份有限公司合成]见表1。

表1 本研究中用到的引物序列Table 1 Oligonucleotide primers used in this study
1.1.2 培养基和缓冲液
(1)培养基 LB 液体培养基(10 g/L 胰蛋白胨,5 g/L 酵母浸出粉,10 g/L NaCl)用于大肠杆菌的常规培养;2×YTPG 培养基(16 g/L 胰蛋白胨,10 g/L酵母浸出粉,5 g/L NaCl,3 g/L KH2PO4,7 g/L K2HPO4,18 g/L 葡萄糖,pH 7.2)用于大肠杆菌细胞提取物制备。
(2)缓冲液 S30 缓冲液[10 mmol/L 三异丙基乙磺酰(Tris)‑醋酸,14 mmol/L 醋酸镁,60 mmol/L 醋酸钾,2 mmol/L 二硫苏糖醇(DTT),pH 8.2]用于细胞提取物制备;蛋白纯化缓冲液(平衡缓冲液:Tris 40 mmol/L,NaCl 500 mmol/L,咪唑0.5 mmol/L,pH 7.5;洗杂缓冲液:Tris 40 mmol/L,NaCl 500 mmol/L,咪唑50 mmol/L,pH 7.5;洗脱缓冲液:Tris 40 mmol/L,NaCl 500 mmol/L,咪唑500 mmol/L,pH 7.5;脱盐缓冲液:Tris 40 mmol/L,NaCl 500 mmol/L,DTT 1 mmol/L,pH 7.5)用于限制性内切酶的纯化。
1.1.3 主要试剂和仪器
(1)试剂 蛋白胨和酵母粉(OXOID),琼脂粉、琼脂糖、ATP、CTP、GTP、UTP、DTT[生工生物工程(上海)],DMSO、异丙基‑β‑D‑硫代半乳糖苷(IPTG)、磷酸烯醇式丙酮酸(PEP)、氧化型辅酶Ⅰ(NAD+)(上海阿拉丁生化科技),氨基酸(Sigma),tRNA(Roche),限制性内切酶BsaⅠ(NEB),限制性内切酶EcoRⅠ、DpnⅠ和BamHⅠ(ThermoFisher Scientific),Phanta Max Super‑Fidelity DNA 聚合酶试剂盒、DNA 回收试剂盒、质粒抽提试剂盒、同源重组克隆试剂盒(南京诺唯赞生物科技)。
(2)仪器 PCR 仪、qPCR 仪、核酸与蛋白质电泳仪(Bio‑Rad),凝胶成像仪(Analytikjena),超声破碎仪(Qsonica),AKTA 蛋白纯化仪(GE),恒温振荡仪与微量分光光度计(杭州奥盛)。
1.2 方法
1.2.1 无细胞表达线性模板的构建
用于无细胞反应的线性模板(linear DNA template,LDT)包括T7 启动子及其上游序列‑融合/纯化标签‑目的基因序列‑T7 终止子及其下游序列(如图1)。其中T7 启动子及其上游序列通过PCR 方法扩增质粒pJL1 或pSUMO 获得,T7 终止子及其下游序列通过扩增质粒pJL1 获得,限制性内切酶基因片段通过扩增质粒pUC‑EcoRⅠ_Kan、pUC‑BamHⅠ_Kan 和pUC‑BsaⅠ_Kan 获得,最后通过融合PCR 方法将3 个片段融合构建线性表达模板,并保证线性模板中不含有目标限制性内切酶的识别位点。融合PCR 反应步骤:10 μL Phanta buffer(5×),1 μL dNTPs(10 mmol/L),3 个片段(约100 ng/片段),ddH2O 补齐至47 μL;98 ℃ 30 s预变性,98 ℃ 10 s,57 ℃ 30 s,72 ℃ 30 s/kb,10个循环,72 ℃ 5 min;引物1/2 各1.5 μL;98 ℃ 30 s预变性,98 ℃ 10 s,57 ℃ 30 s,72 ℃ 30 s/kb,30个循环,72 ℃ 5 min。PCR产物经切胶回收浓缩后备用。
1.2.2 无细胞提取物的制备
以E. coliBL21 Star(DE3)菌株为宿主,活化后挑取单菌落于20 mL 的LB 培养基中,于32 ℃、220 r/min过夜培养;以初始OD600≈0.05的接种量转接至1 L 2×YPTG培养基中,于34 ℃、220 r/min培养至OD600≈0.8,添加0.5 mmol/L 的IPTG 诱导T7 RNA 聚合酶表达,继续培养2.5~3 h 至OD600>2.5;于4 ℃、5000g离心15 min,收集菌体,用S30 缓冲液重悬洗涤菌体3次,得到菌体。
菌体称重,1 g 菌体加入1 mL S30 缓冲液,涡旋重悬,得到菌悬液;1.4 mL 菌悬液置于1.5 mL离心管中超声破碎,破碎程序为:50%功率,10 s超声10 s 暂停,至能量达到600~620 J;然后于4 ℃、12 000g离心10 min,取上清;上清于4 ℃、10 000g离心10 min,获得的上清即为细胞提取物。将细胞提取物于-80 ℃冻存备用。
1.2.3 限制性内切酶的无细胞表达和纯化
(1)无细胞表达限制性内切酶体系为(15 μL)12 mmol/L 谷氨酸镁,10 mmol/L 谷氨酸铵,130 mmol/L 谷氨酸钾,1.2 mmol/L 三磷酸腺苷(ATP),0.85 mmol/L三磷酸鸟苷(GTP),0.85 mmol/L三磷酸尿苷(UTP),0.85 mmol/L 三磷酸胞苷(CTP),34 μg/mL亚叶酸,170 μg/mL大肠杆菌tRNA混合物,2 mmol/L 20种必需氨基酸,0.33 mmol/L烟酰胺腺嘌呤二核苷酸(NAD),0.27 mmol/L 辅酶A(CoA),1.5 mmol/L 亚精胺,1 mmol/L 腐胺,4 mmol/L 草酸钠,33 mmol/L 磷酸烯醇式丙酮酸(PEP),5 μmol/L 线性表达模板,4 μL 细胞提取物。反应体系在1.5 mL 无酶离心管中于30 ℃静置表达6 h。
(2)产物检测 反应体系不离心样品为全菌表达蛋白(total,T),反应体系于13 000g离心10 min取上清,为可溶表达蛋白(soluble,S)。反应产物经蛋白质印迹法(Western blot)检测表达情况,其中一抗为6*His‑标签鼠单克隆抗体。
(3)产物纯化 等比例扩大无细胞反应体系至4 mL,反应体系在50 mL 离心管中于30 ℃振荡(250 r/min)表达6 h。反应结束后,反应体系于4 ℃、21 000g离心40 min,取上清,加入等体积的平衡缓冲液,进行Ni‑NTA 亲和色谱纯化(Superflow Agarose,1 mL 预装柱,GE),10 mL洗杂缓冲液去除未结合的杂蛋白,2 mL 洗脱缓冲液洗脱收集目标蛋白,以聚丙烯酰胺凝胶电泳(SDS‑PAGE)检测纯化产物。目标蛋白经脱盐浓缩(超滤管,分子截留10~30 kDa)至1 mL,测定吸光度A280nm计算蛋白浓度。目标蛋白经凝胶色谱方法进一步纯化(柱型号:Hiload 26/600 superdex 75 pg),以脱盐缓冲液进行等度洗脱,洗脱后的样品经SDS‑PAGE 验证纯度,浓缩后测定280 nm 下的吸光度,结合目标蛋白的摩尔消光系数(A1mg/mL280nm)确定最终的蛋白浓度,向蛋白样品中补充甘油至终浓度为10%,于-80 ℃保存备用。
1.2.4 限制性内切酶的活性检测
(1)实时酶活检测 以pJL1‑sfGFP质粒为基础,构建sfGFP表达盒区域(T7启动子至T7终止子区)含有目标限制性内切酶识别位点的质粒作为实时酶活检测底物。在15 μL 无细胞反应体系中共表达目标限制性内切酶和对应的sfGFP质粒,以不含限制性内切酶表达模板的反应为阴性对照,在qPCR仪中反应(30 ℃)并实时检测sfGFP的产量(即荧光信号)。限制性内切酶在无细胞体系中成功表达后,有活性的酶蛋白即会识别相应酶切位点对底物DNA进行切割,导致sfGFP的表达模板被破坏,使得sfGFP产量不再增加,荧光值趋于平稳。反应过程中实验组和对照组sfGFP荧光值的变化即可反映无细胞表达目标限制性内切酶的活性。
(2)酶活测定 根据限制性内切酶酶活定义,1 U表示在37 ℃下,总体积为20 μL的反应体系中,能够在1 h内消化1 μg的质粒DNA(含有1个相应限制性内切酶位点)所需的酶量。选择或构建含有目标限制性内切酶识别序列的质粒作为酶活测定底物,以脱盐缓冲液梯度稀释成不同浓度的目标限制性内切酶为实验组,以商业化限制性内切酶为阳性对照,反应体系为(20 μL):2 μL 10×Buffer(ThermoFisher Scientific),5 μL 梯度稀释后目标限制性内切酶(阳性对照添加1 μL商业化酶),1 μg底物质粒,ddH2O 补足体系至20 μL。反应条件为:37 ℃,1 h。反应结束后取样进行琼脂糖核酸电泳,根据酶切结果判定目标限制性内切酶的比活。
2 结果与讨论
2.1 线性模板构建
基因模板是无细胞蛋白表达最重要的组分之一,研究表明环状质粒、线性DNA 片段和mRNA序列均可作为模板用于目的蛋白的体外表达,其中质粒因其稳定性好、制备简便等优点,是CFPS中首选的模板形式。大肠杆菌无细胞表达体系常用的T7启动子在克隆宿主E. coliDH5α中有一定的泄露表达,而限制性内切酶对DNA 序列的切割活性较高,使得T7 系列启动子的泄露表达足以对宿主细胞造成一定的毒性,从而影响限制性内切酶质粒模板的构建和扩增。因此,本研究选择线性DNA 片段作为表达模板,可以适用于表达水平或酶切活性有差异的所有限制性内切酶。为了提高线性模板在无细胞体系中的稳定性,在模板表达盒(expression cassette)上下游各增加约300 bp 序列,以降低无细胞体系中核酸酶对线性模板降解的影响。以限制性内切酶EcoRⅠ为例(LDT‑EcoRⅠ),在基因编码区N 端添加6*His‑tag 作为纯化标签,选择性添加融合蛋白如SUMO 以促进可溶表达,添加蛋白酶酶切位点如HRV 3C 以去除N 端标签[示意图见图1“(1)线性模板构建”],表达盒内所有序列以突变方式优化去除EcoRⅠ的酶切位点。
2.2 无细胞表达限制性内切酶EcoRⅠ
EcoRⅠ的无细胞表达基于常用的E. coliBL21 Star(DE3)无细胞提取物。当以线性片段为表达模板时可以获得大量的目的蛋白,但可溶性较差[不足10%的可溶表达,见图2(a)]。随后对表达模板和无细胞体系进行了优化,结果发现,在含有促可溶分子伴侣(DnaK‑DnaJ‑GrpE,GroES‑GroEL,pG‑KJE8)的体系中目的蛋白的可溶性有所提高(约30%的可溶表达)。为了进一步提高目的蛋白的可溶表达,选择融合标签蛋白SUMO(small ubiquitin‑related modifier),SUMO 蛋白来源于真核细胞酿酒酵母翻译后修饰系统中的Smt3 蛋白,常以N 端融合标签促进原核生物中目的蛋白的异源表达。SUMO 等泛素蛋白具有快速折叠的结构[34],快速折叠完成的SUMO 蛋白为C 端融合的目的蛋白提供了折叠成核位点[35‑36],引导并促进目的蛋白的正确折叠,从而提高目的蛋白的可溶表达。SUMO 融合标签促可溶能力强[37],且与EcoRⅠ蛋白分子量相差适宜(20 kDa),便于进一步分离纯化,因此本部分实验选用SUMO标签融合EcoRⅠ在无细胞体系中表达。由于SUMO蛋白编码序列中包含有一个EcoRⅠ的酶切位点,为避免表达产物对模板的降解,通过同义密码子优化去除此酶切位点(R71,aga→cgt)后构建表达模板。结果表明,以N端融合SUMO标签的线性片段作为表达模板,在BL21 Star(DE3)无细胞体系中,SUMO‑EcoRⅠ可以实现100%的可溶表达[图2(b)]。

图2 限制性内切酶EcoRⅠ的无细胞制备(Western blot和SDS‑PAGE检测无细胞表达、纯化的限制性内切酶EcoRⅠ)CE-cell extract,以BL21 Star (DE3)制备的细胞提取物;CE+ -含有分子伴侣(pG‑KJE8)的BL21 Star(DE3)细胞提取物;T-全菌蛋白;S-可溶蛋白;M-蛋白标准样品;FT-纯化上样穿出液;W-洗杂流出液;Eluate-洗脱液Fig. 2 Cell‑free production of restriction endonuclease EcoRⅠ(Western blot and SDS‑PAGE analyses of cell‑free expressed and purified restriction endonuclease EcoRⅠ)CE-cell extract of BL21 Star (DE3); CE+ -cell extract of BL21 Star (DE3) with chaperone (pG‑KJE8); T-total protein;S-soluble protein; M-marker; FT-flow throughout sample; W-washed sample; Eluate-eluted sample
为了检测无细胞表达EcoRⅠ的产量并进一步验证其催化活性,将无细胞表达体系由15 μL 扩大至4 mL,并对表达产物进行分离纯化。如图2 所示,经亲和色谱纯化后目的蛋白[SUMO‑EcoRⅠ,45.4 kDa,见图2(c)]浓度约为1 mg/mL(体积为1 mL)。经蛋白酶HRV 3C 酶切去除SUMO 标签后[图2(d)],进一步进行凝胶色谱纯化,得到最终目的蛋白EcoRⅠ[32.0 kDa,0.54 mg/mL,1 mL,纯度约95%,见图2(e)]。
2.3 无细胞制备限制性内切酶的酶活分析
为了验证无细胞表达的限制性内切酶是否有切割DNA 的活性,我们建立了基于无细胞反应的实时酶活检测方法(见1.2.4实时酶活检测)。以含有EcoRⅠ酶切位点的质粒pSUMO‑sfGFP_EcoRⅠ为底物(该质粒中的SUMO 蛋白编码区含有EcoRⅠ酶切位点,未突变),在无细胞体系中共表达sfGFP 和SUMO‑EcoRⅠ。如图3 所示,无细胞反应进行约1h 后,阴性对照组中sfGFP 的产量持续上升,而实验组由于SUMO‑EcoRⅠ积累了一定量的活性蛋白,开始催化切割质粒底物,导致表达模板被破坏,sfGFP 的产量上升非常缓慢;反应进行6 h 后实验组sfGFP 的产量趋于平稳。这表明无细胞表达的SUMO‑EcoRⅠ在30 ℃的无细胞体系中具有切割底物DNA 的活性,SUMO‑EcoRⅠ蛋白合成后迅速切割质粒底物(1 h 以内),随着SUMO‑EcoRⅠ蛋白的积累,底物被彻底切割,sfGFP 的终产量显著低于对照组。这一实时酶活检测方法具有反应体系小(15 μL)、操作简便(无需纯化目的酶蛋白)、反应条件灵活(如温度适用23~37 ℃)、响应灵敏(3 h 以内)、通量高(96 孔板)等优势,将有助于促进限制性内切酶的挖掘、改造、进化和活性筛选等方面的研究。
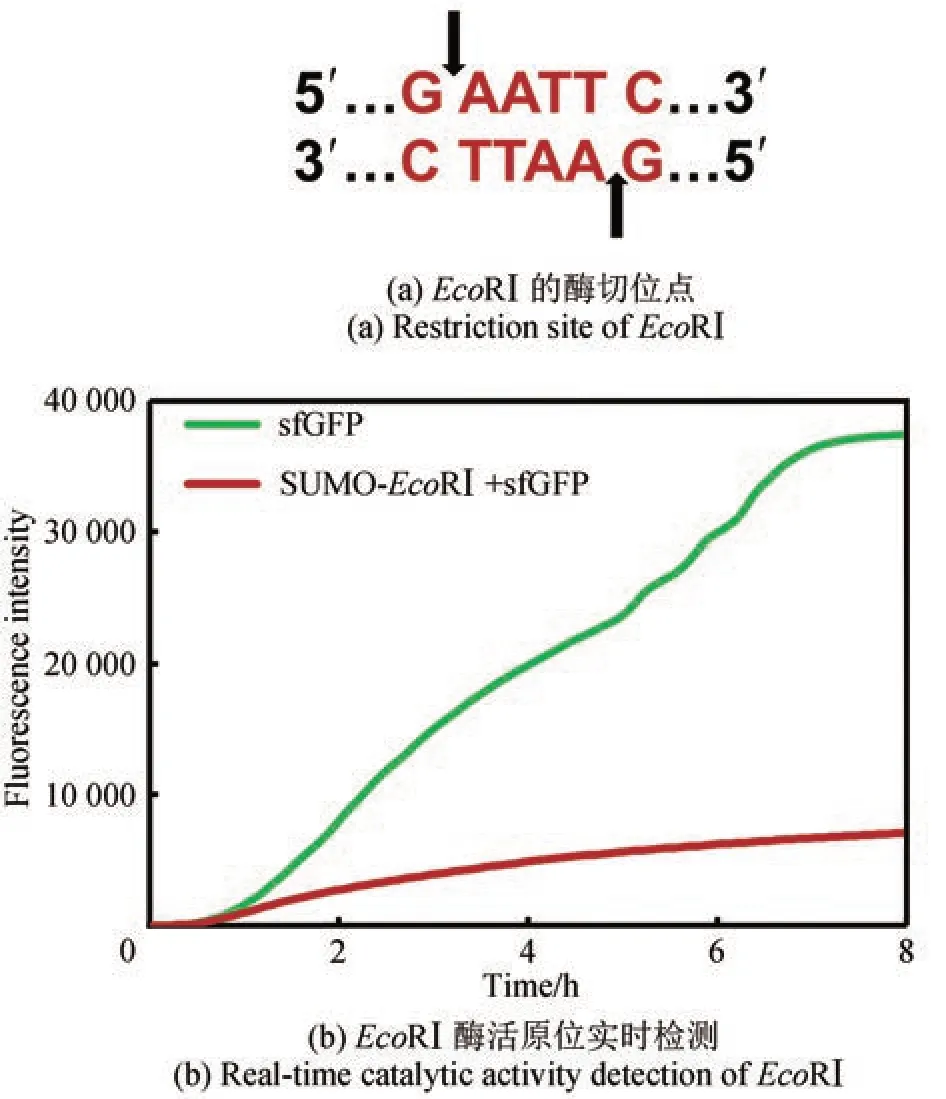
图3 无细胞体系中限制性内切酶EcoRⅠ酶活的实时检测Fig. 3 Real‑time catalytic activity detection of restriction endonuclease EcoRⅠ in CFPS system
根据限制性内切酶酶活定义,将无细胞制备的EcoRⅠ进行梯度稀释为0.54×10-1mg/mL、0.54 ×10-2mg/mL、0.54×10-3mg/mL 和0.54×10-4mg/mL,以空质粒pRSFDuet‑1(1 μg)为底物,以商业化EcoRⅠ(ThermoFisher Scientific)为阳性对照进行酶切反应。结果如图4,无细胞制备的EcoRⅠ可以实现底物DNA 分子的识别和切割,随着酶浓度的降低,切割活力有所减弱,当酶浓度大于等于0.54×10-2mg/mL 时可以将底物彻底切割消化,酶浓度降低至0.54×10-3mg/mL 时,有少量底物残余。据此计算无细胞制备EcoRⅠ的酶活为3.7×105~3.7×106U/mg。
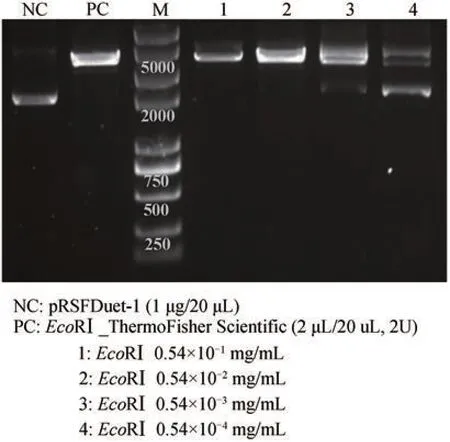
图4 无细胞制备限制性内切酶EcoRⅠ的酶活测定NC-阴性对照;PC-阳性对照Fig. 4 Catalytic activity determination of cell‑free produced restriction endonuclease EcoRⅠNC-negative control; PC-positive control
2.4 无细胞制备限制性内切酶BamHⅠ和BsaⅠ
为了进一步验证我们构建的限制性内切酶无细胞制备方法是否适用于更多限制性内切酶的表达,又选择了BamHⅠ和BsaⅠ进行无细胞制备。经无细胞体系表达后,结果显示,BamHⅠ和BsaⅠ不需要N 端融合促可溶标签SUMO,也不需要含有促可溶分子伴侣的无细胞提取物,仅带有简单的纯化标签6*His‑tag,在E. coliBL21 Star(DE3)无细胞体系中即可表达出可溶的目的蛋白[图5(a)和(d)]。分别以含有相应酶切位点的质粒pJL1‑sfGFP_BamHⅠ和pJL1‑sfGFP_BsaⅠ(酶切位点位于T7 启动子之后)作为底物和线性模板LDT‑BamHⅠ和LDT‑BsaⅠ共表达,sfGFP 实时荧光检测结果表明,无细胞表达的6*His-BamHⅠ和6*His-BsaⅠ在无细胞体系中均有底物切割活性[图6(c)和(d)]。扩大无细胞体系至4 mL 大量表达,并经过亲和色谱和凝胶色谱两步纯化后,均获得正确大小的目的蛋白BamHⅠ(25.4 kDa)0.25 mg/mL(体积为0.5 mL)、BsaⅠ(64.4 kDa)0.45 mg/mL(体积为1 mL)[图5(b)、(c)、(e)和(f)]。以商业化酶BamHⅠ(ThermoFisher Scientific)和BsaⅠ(NEB)为阳性对照进行酶活测定,结果显示无细胞制备的BamHⅠ和BsaⅠ均有切割底物的活性[图6(e)和(f)],酶活分别为BamHⅠ 8.3×102~4.1×103U/mg和BsaⅠ 4.4×105~4.4×106U/mg。其中BamHⅠ的表达量和酶活相对较低,这主要是由于BamHⅠ纯化后的产量较低、终浓度较低,需要对BamHⅠ的无细胞表达体系和后续纯化步骤做进一步的调整优化,如改进模板序列组成、提高模板浓度以提高蛋白表达量,优化纯化方法中的缓冲液组分、凝胶色谱条件等以减少纯化过程中的损失,最终提高无细胞制备终产物的产量,提高酶活。

图5 限制性内切酶BamHⅠ和BsaⅠ的无细胞制备(Western blot和SDS‑PAGE检测无细胞表达纯化的限制性内切酶BamHⅠ和BsaⅠ)Fig. 5 Cell‑free production of restriction endonucleases BamHⅠ and BsaⅠ(Western Blot and SDS‑PAGE analyses of cell‑free expressed and purified restriction endonucleases BamHⅠ and BsaⅠ)
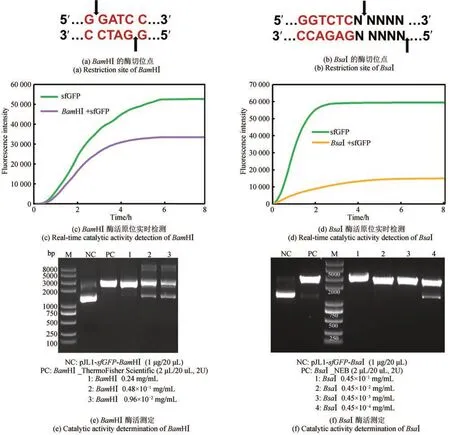
图6 无细胞制备限制性内切酶BamHⅠ和BsaⅠ的酶活分析Fig. 6 Catalytic activity assay of cell‑free produced restriction endonucleases BamHⅠ and BsaⅠ
综上,经无细胞制备的限制性内切酶纯度较高(大于95%),产量可达0.03~0.13 mg/mL 无细胞反应(32.5~130 mg/L 无细胞反应),酶活可达4.1×103~4.4×106U/mg。相较于传统技术制备的限制性内切酶,如BsaⅠ(产量为0.6~10.2 mg/L 重组表达,酶活为2.5×105U/mg)[16],SalⅠ(产量为10 mg/L重组表达,酶活为4×106U/mg)[17],KpnⅠ(产量为1.28×105U/L 重组表达,酶活为1.28×105U/mg)[38],NcoⅠ(产量为10 mg/L 重组表达,酶活为6×106U/mg)[39],无细胞方法制备的限制性内切酶酶活相当,且产量较高。
3 结 论
本研究基于无细胞蛋白表达技术开发建立了限制性内切酶的无细胞制备系统,即利用大肠杆菌无细胞体系进行限制性内切酶的体外快速合成、酶活检测和纯化制备。本研究首先对限制性内切酶EcoRⅠ进行了无细胞合成与制备,结果显示,在没有甲基化酶的辅助下,EcoRⅠ可以在无细胞体系中成功表达、纯化,并具有酶切DNA活力;随后将此方法应用于另外两种限制性内切酶BamHⅠ和BsaⅠ,也成功实现了这两个酶的无细胞制备与活性测定。总的来说,这一限制性内切酶的无细胞制备体系具有操作简单、生产周期短(1~2 天/制备)、蛋白产量高(32.5~130 mg/L无细胞反应)、制备效率高(1.3×105~5.7×108U/L无细胞反应)等优势,这将对限制性内切酶的发掘、研究、制备和商业化推广等方面具有良好的应用前景。
猜你喜欢
云南化工(2021年6期)2021-12-21
中学生数理化·中考版(2021年9期)2021-11-20
中国科技纵横(2021年24期)2021-03-02
科学(2020年2期)2020-08-24
中成药(2018年6期)2018-07-11
临床医药文献杂志(电子版)(2017年11期)2017-05-17
临床骨科杂志(2017年1期)2017-03-07
生物技术通报(2015年1期)2015-04-10
山西大同大学学报(自然科学版)(2013年5期)2013-09-13
化学分析计量(2013年3期)2013-03-11
