
阿普斯特片在健康受试者中的生物等效性研究
2023-09-26 05:47程玲秦月雯杨艺茜王勇李剑徐满
现代药物与临床 2023年8期
程玲,秦月雯,杨艺茜,王勇,李剑,徐满*
1.江西青峰药业有限公司,江西 赣州 341000
2.南昌大学第二附属医院,江西 南昌 330000
阿普斯特是一种新型的口服小分子磷酸二酯酶4(PDE4)抑制剂,可抑制PDE4 活性,提高细胞内环磷酸腺苷水平,进一步调控肿瘤坏死因子和其他炎性细胞因子的表达,最终抑制炎症反应[1-2]。临床数据显示,阿普斯特能够显著改善指甲、头皮、掌跖(手足)的银屑病,有效治疗银屑病关节炎,为银屑病患者群体提供了一种非常有价值的治疗选择[3]。2019 年7 月,阿普斯特获得了美国食品药品管理局批准,用于治疗与白塞氏病相关的口腔溃疡[4]。阿普斯特口服吸收良好,绝对生物利用度为73%[5]。在约2.5 h 的达峰时间(tmax)出现峰浓度(Cmax),Cmax约为584 ng/mL[6],食物不影响阿普斯特的吸收。血浆蛋白结合率约为68%,平均表观分布容积(Vd)为87 L[5]。口服给药后,阿普斯特(45%)为体内循环主要成分,其次为非活性代谢物M12(39%)。阿普斯特可在人体内广泛代谢,在血浆、尿液和粪便中已发现23 种代谢产物[7]。阿普斯特通过细胞色素(CYP)氧化代谢、随后通过葡糖苷酸化和非CYP 介导的水解作用进行代谢。在体外,阿普斯特的CYP 代谢主要由CYP3A4 介导,CYP1A2、CYP2A6 的作用较小。在健康受试者中,阿普斯特的血浆清除率约为10 L/h,终末消除半衰期为6~9 h。放射性标记的阿普斯特经口给药后,以阿普斯特形式存在的放射性剂量在尿液和粪便中分别占3%、7%[5]。本研究采用高效液相色谱-质谱(HPLC-MS/MS)法测定健康受试者在空腹和餐后状态下服用30 mg 阿普斯特片后人血浆中阿普斯特的质量浓度,考察国产阿普斯特片和原研药在健康受试者中的药动学特征和生物等效性。
1 材料与对象
1.1 药品
受试制剂(T):阿普斯特片,江西青峰药业有限公司生产,规格30 mg/片,批号2020 111003;参比制剂(R):阿普斯特片,商品名Otezla®,Amgen Europe B.V.生产,规格30 mg/片,批号H02445A。阿普斯特对照品,质量分数98.9%,批号2314-033A2,TLC Pharmaceutical Standards ;内 标Apremilast-d5对照品,质量分数96.4%,批号2314-040A3,TLC Pharmaceutical Standards。
1.2 仪器
LC-20ADXR 高效液相色谱仪(Shimadzu 公司),LC-20ADXR 高效液相色谱泵(Shimadzu 公司),Turbo Spray 离子源(AB Sciex),SIL-30ACMP自动进样器(Shimadzu 公司),CTO-20AC 柱温箱(Shimadzu 公司);Triple Quad 4500 质谱仪(AB Sciex 公司);XPR6UD5 千万分之五电子天平[梅特勒-托利多仪器(上海)有限公司];Sorvall Legend Micro 17R 高速离心机(Thermo Scientific);Multifuge X1R 高速离心机(Thermo Fisher);MTV-100 涡旋混合仪(杭州奥盛仪器有限公司),MX-M涡旋混合仪[大龙兴创实验仪器(北京)有限公司];移液站:OL7001-26-214,CyBi SELMA 96,德国耶拿分析仪器股份公司;Analyst 1.6.3 分析软件(AB Sciex)。
1.3 受试者的选择
根据研究报道[8-9],阿普斯特片参比制剂空腹Cmax个体内变异度小于30%,为22.5%。假设α=0.05,β=0.2,几何均值比GMR=95%、100%、105%,CV 为24%,脱落率为20%,生物等效区间为80.00%~125.00%,采用SAS 9.4 软件估计空腹和餐后样本量分别为32 例。
本研究经南昌大学第二附属医院医学伦理委员会批准,批件号:药临审[2021]第(22)号。所有受试者均自愿签订知情同意书。空腹试验和餐后试验分别入组32 例合格健康受试者,随机分为T-R(受试制剂/参比制剂)和R-T(参比制剂/受试制剂)组,每组各16 例受试者。
入选标准:18~50 周岁男性和女性健康受试者,BMI 在19.0~26.0 kg/m2,男性受试者≥50.0 kg,女性受试者≥45.0 kg。
排除标准:3 个月内参加过其他的药物临床试验者或接受外科手术;有药片吞咽困难者;采血困难者;对阿普斯特、辅料中成分过敏者或过敏体质;嗜烟嗜酒者;3 个月内献血或大量失血者;胃肠疾病史者;既往出现过心律失常状况者;30 d 内使用过与阿普斯特有相互作用的药物;哺乳期或妊娠期女性;有药物滥用史或药物滥用筛查阳性者;30 d内使用过抑制或诱导肝脏对药物代谢的药物。
空腹试验组共筛选受试者64 例,成功入组32例。空腹试验入组男性24 例,女性8 例,年龄为(25.94±4.66)岁,平均身高为(167.81±7.33)cm,平均体质量为(62.42±6.36)kg,平均体质量指数(BMI)为(22.15±1.62)kg/m2。空腹试验过程中,2 例受试者脱落,另1 例受试者第2 周期数据被剔除(给药后首个样品为Cmax,且未采集给药后5~15 min 的早期样品)。餐后试验组共筛选79 例,成功入组32 例。餐后试验入组男性24 例,女性8 例,年龄为(20.78±2.52)岁,平均身高为(168.58±7.00)cm,平均体质量为(63.37±7.92)kg,平均BMI 为(22.24±1.87)kg/m2。餐后试验过程中,1例受试者发生脱落,无受试者被剔除。空腹试验32例受试者均纳入安全性分析集,31 例纳入药动学参数集,30 例纳入生物等效性分析集。餐后试验32例受试者全部纳入安全性分析集、药动学参数集和生物等效性分析集。
2 方法与结果
2.1 给药和样本采集
本研究采用单中心、随机、开放、两制剂、两序列、两周期交叉的生物等效性试验设计。筛选期选择合格的健康成年受试者随机至T-R 组和R-T组,按顺序交叉给药。参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》[10]的要求,设置清洗期7 d。筛选合格的受试者于试验前1 d 入住。服药前1 h 至服药后1 h 内禁止饮水,给药后禁食4 h,4 h 后进食标准午餐,10 h 后进食标准晚餐。
空腹试验受试者隔夜空腹至少10 h 以上,给药当天早上按随机表服用受试制剂或参比制剂,每次1 片,30 mg/片,以240 mL 温水送服。每个周期分别于给药前0 h(给药前1.0 h 内)和给药后20、40 min 以及1.0、1.5、2.0、2.5、3.0、3.5、4.0、6.0、8.0、12.0、24.0、48.0 h 采集上肢静脉血约4 mL。
餐后试验受试者隔夜空腹至少10 h 后,在给药前30 min 开始进食高脂餐,控制进餐速度,要在30 min 内吃完,药物要在进餐后且以开始进餐时间计的第30 分钟服用,按随机表服用受试制剂或参比制剂,1 片/次,30 mg/片,以240 mL 温水送服。受试者于给药前0 h(给药前1.0 h 内)和给药后0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0、5.0、6.0、8.0、12.0、24.0、48.0 h 采集上肢静脉血,每次取血4 mL。
所得全血样品在采血后1 h 内放进4 ℃低温离心机(2~8 ℃,1 700×g)离心10 min,,然后分离血浆样品。所有离心后血浆样品分成两份,至少取0.8 mL 血浆加入到检测冻存管中,剩余血浆样品放入到备份冻存管中,血浆样品分装在冰浴下进行。血浆样品需在采集后2 h 内置于-20 ℃冰箱暂存,并在采血后24 h 内转运至-80 ℃超低温冰箱(-60~-90 ℃)进行冻存,或在样本离心操作完成2 h 内将分装后的血浆直接转移至温度-80 ℃超低温冰箱(-60~-90 ℃)保存。
2.2 HPLC-MS/MS 法测定阿普斯特
2.2.1 色谱条件 Ultimate XB C18色谱柱(50.0 mm×2.1 mm,5 μm);流动相:含0.01%甲酸的水溶液(A)-乙腈(B);梯度洗脱,0~0.10 min 35.0%B,0.10~1.20 min 35.0%~55.0% B,1.20~1.30 min 55.0%~90.0% B,1.30~2.00 min 90.0% B,2.00~2.10 min 90.0%~35.0% B;体积流量1.0 mL/min;进样体积7.00 μL;柱温35 ℃。
2.2.2 质谱条件 ESI 电喷雾离子源,正离子模式,MRM 多反应监测扫描,涡旋离子喷雾温度600.00 ℃,电喷雾电压5 500.00 V,气帘气30.00 psi(1 psi=6 895 Pa),雾化气50.00 psi,辅助气50.00 psi,入口电压:10.00 V,碰撞室出口电压:10.00 V。阿普斯特m/z461.1→257.1,去簇电压100.00 V,碰撞能量15 eV,阿普斯特-d5466.2→262.1,去簇电压90.00 V,碰撞能量16.00 eV。
2.2.3 溶液的制备
(1)阿普斯特储备液的制备:取一定量的阿普斯特2 份,置于透明玻璃瓶中,记录质量。根据其质量分数、水分、是否成盐等信息计算该分析物的真实质量,加入适量的甲醇-二甲基亚砜(95∶5),涡旋混合,配制0.500 mg/mL 的储备液。
(2)标准曲线工作溶液的制备:在透明玻璃瓶中加入一定量90%甲醇溶液,然后加入阿普斯特储备液,涡旋混合,配制成最终质量浓度分别为50、100、400、2 000、5 000、10 000、18 000、20 000 ng/mL 的工作液。
(3)内标储备液的制备:根据阿普斯特-d5质量分数、水分、是否成盐等信息,将1.04 mg 内标对照品加入0.997 mL 甲醇-二甲基亚砜(95∶5),涡旋混合,配制成阿普斯特-d5最终质量浓度为1.00 mg/mL 的储备液。
(4)内标工作溶液的制备:在透明玻璃瓶中将内标储备液用90%甲醇溶液稀释为5 000 ng/mL 的内标工作溶液,将5 000 ng/mL 内标工作溶液用90%甲醇溶液稀释为100 ng/mL 内标工作溶液。
(5)标准曲线溶液的制备:在透明聚丙烯离心管中加入适量的混合空白血浆(高脂效应所在分析批使用混合空白正常血浆),然后加入相应质量浓度的标准曲线工作溶液,涡旋混匀,制成质量浓度分别为2.00、4.00、16.00、80.00、200.00、400.00、720.00、800.00 ng/mL 的校正标样。
(6)质控工作溶液和质控样品溶液的制备:在透明玻璃瓶中加入一定量90%甲醇溶液,然后加入一定体积阿普斯特储备液,涡旋混合,制成50、150、1 500、6 000、15 000、80 000 ng/mL 的质控工作溶液。在透明聚丙烯离心管中加入适量的混合空白血浆,加入相应质量浓度工作溶液,涡旋混匀,得2.00(LLOQ QC)、6.00(LQC)、60.00(MQC2)、240.00(MQC)、600.00(HQC)、3 200.00(DQC)ng/mL的质控样品溶液。
2.2.4 血浆样品处理 采用乙腈蛋白沉淀法。在一块96 孔深孔板中分别往空白基质样品、零浓度样品的孔位中加入50.0 μL 空白血浆,空白试剂样品的孔位中加入同等体积的超纯水,校正标样的孔位中加入同等体积的校正标样,质控样品的孔位中加入同等体积的质控样品,不加内标的定量上限样品(ULOQ-no IS)样品的孔位中加入同等体积的定量上限样品(其中稀释质控样品先用适量的混合空白血浆稀释);往空白基质样品[包括基质效应评价样品(MER)和回收率评价样品(REC))、空白试剂样品(包括基质效应评价的纯溶液样品(MEP)]和ULOQ-no IS 样品的孔位中分别加入25.0 μL 90%甲醇溶液,往其他样品的孔位中加入同等体积的内标工作溶液,涡旋混合3 min;以上操作全程需在湿冰上进行。使用移液器/移液站给每个样品加入400 μL乙腈,涡旋混合5 min;在4 ℃的高速冷冻离心机以1 700×g离心15 min;转移100 μL 上清液至另一块200 μL 含1%甲酸的50%甲醇溶液的96 孔深孔板中,REC、MEP、MER 样品加入200 μL 低(LRS)、中(MRS)、高浓度(HRS)回收率样品溶液,涡旋混合5 min。在进样分析前将样品处理后的提取物保存在自动进样器的温度下或4 ℃的冰箱中。
2.2.5 选择性试验 6 个不同来源的空白正常基质、1 个来源的空白高脂基质、1 个来源的空白溶血基质对分析物和内标无明显干扰,不影响定量分析。混合来源的空白基质(MTX-BLK)对分析物和内标的检测无明显干扰,不影响定量分析。空白试剂溶液(RCT-BLK)对分析物和内标的检测无明显干扰,不影响定量分析。内标对分析物、分析物对内标的检测无明显干扰,不影响定量分析。同位素内标短期室温放置24 h 对分析物的检测无明显干扰,不影响定量分析。同位素内标在4 ℃储存条件下放置38 d 对分析物的检测无明显干扰,不影响定量分析。见图1。

图1 HPLC-MS/MS 色谱图Fig.1 HPLC-MS/MS chromatograms
2.2.6 标准曲线方程和定量下限 对理论质量浓度与响应值进行线性回归,采用最小二乘法对标准曲线所有质量浓度点进行拟合,得方程Y=0.025 6X-0.000 367(R2=0.998 1)。阿普斯特的线性范围为0.20~200.00 ng/mL,定量下限为0.200 ng/mL。
2.2.7 准确度和精密度试验 配制质量浓度分别为2.00、6.00、60.00、240.00、600.00 ng/mL 的质控样品LLOQ QC、LQC、MQC2、MQC 和HQC,进样测定,结果见表1。结果表明批内、批间准确度和精密度良好。
表1 精密度和准确度试验结果(,n=3)Table 1 Results of precision and accuracy tests (,n=3)

表1 精密度和准确度试验结果(,n=3)Table 1 Results of precision and accuracy tests (,n=3)
2.2.8 稳定性试验 采用低浓度质控样品(6.00 ng/mL)和高浓度质控样品(600.00 ng/mL)分别测定稳定性。血浆中分析物在湿冰放置24 h;处理过的样品在自动进样器4 ℃下存储100 h;在-20、-80 ℃条件下经过5 次冻融;血浆中分析物储存在-20 ℃条件下26 d;在-80 ℃条件下59 d,结果见表2。
表2 稳定性试验结果(,n=3)Table 2 Results of stability test (,n=3)

表2 稳定性试验结果(,n=3)Table 2 Results of stability test (,n=3)
2.2.9 提取回收率试验 用混合空白基质配制6.00、240.00、600.00 ng/mL 3 个质量浓度的质控样品,混合空白基质处理后加入与6.00、240.00、600.00 ng/mL 相同质量浓度的分析物溶液,配制成回收率评价样品,每一个质量浓度平行操作6 份,进行测定,计算提取回收率,结果平均提取回收率分别为101.6%、101.2%、101.6%,RSD 值分别为2.1%、2.5%、1.8%;内标平均提取回收率分别为108.9%、105.2%、107.7%,RSD 值分别为1.3%、1.5%、1.4%。
2.2.10 基质效应 6 个不同来源的空白正常基质、1 个来源的空白高脂基质、1 个来源的空白溶血基质对分析物和内标的基质效应接近,不影响分析物的定量分析。低(LQC)、高(HQC)质量浓度的样品经内标归一化的基质因子的平均值分别为0.986、1.010,RSD 值分别为5.8%、1.4%。溶血效应中LQC和HQC 的RSD 值分别为6.3%、3.3%,平均准确度偏差为-5.4%、3.4%,无明显溶血效应,不影响定量分析。高脂效应中LQC、HQC 的RSD 值分别为2.2%、2.3%,平均准确度偏差为-3.8%、3.4%,无明显高脂效应,不影响定量分析。
2.2.11 残留 批量样品检测结果发现分析物残留率为0~3.0%,内标无残留,分析物、内标残留均可忽略。
2.2.12 稀释可靠性 用空白基质将DQC 样品稀释5 倍,平行操作6 份,RSD 值为2.2%,表明当稀释度不大于1∶5 时,用空白基质稀释样品对测定结果影响不大。
2.3 药动学数据和统计分析
血药浓度数据采用Phoenix WinNonlin软件(8.2版本)进行药动学参数的计算,根据每个受试者的个体血药浓度,采用非房室模型(NCA)计算阿普斯特的药动学参数,包括Cmax、AUC0-t、AUC0-∞、tmax、t1/2、λz、AUC_%Extrap。采用BES,将受试制剂和参比制剂Cmax、AUC0-t和AUC0-∞经对数转换后进行方差分析(ANOVA)。方差分析模型中给药顺序、药物、周期作为固定效应,受试者(顺序)作为随机效应。计算主要指标的几何均值比率(受试制剂/参比制剂)的90%置信区间,并进行等效性比较,若Cmax、AUC0-t和AUC0-∞几何均值比的90%置信区间落在80.00%~125.00%,则认为两种制剂生物等效。
空腹和餐后试验受试者口服受试制剂和参比制剂的血药浓度-时间曲线见图2。
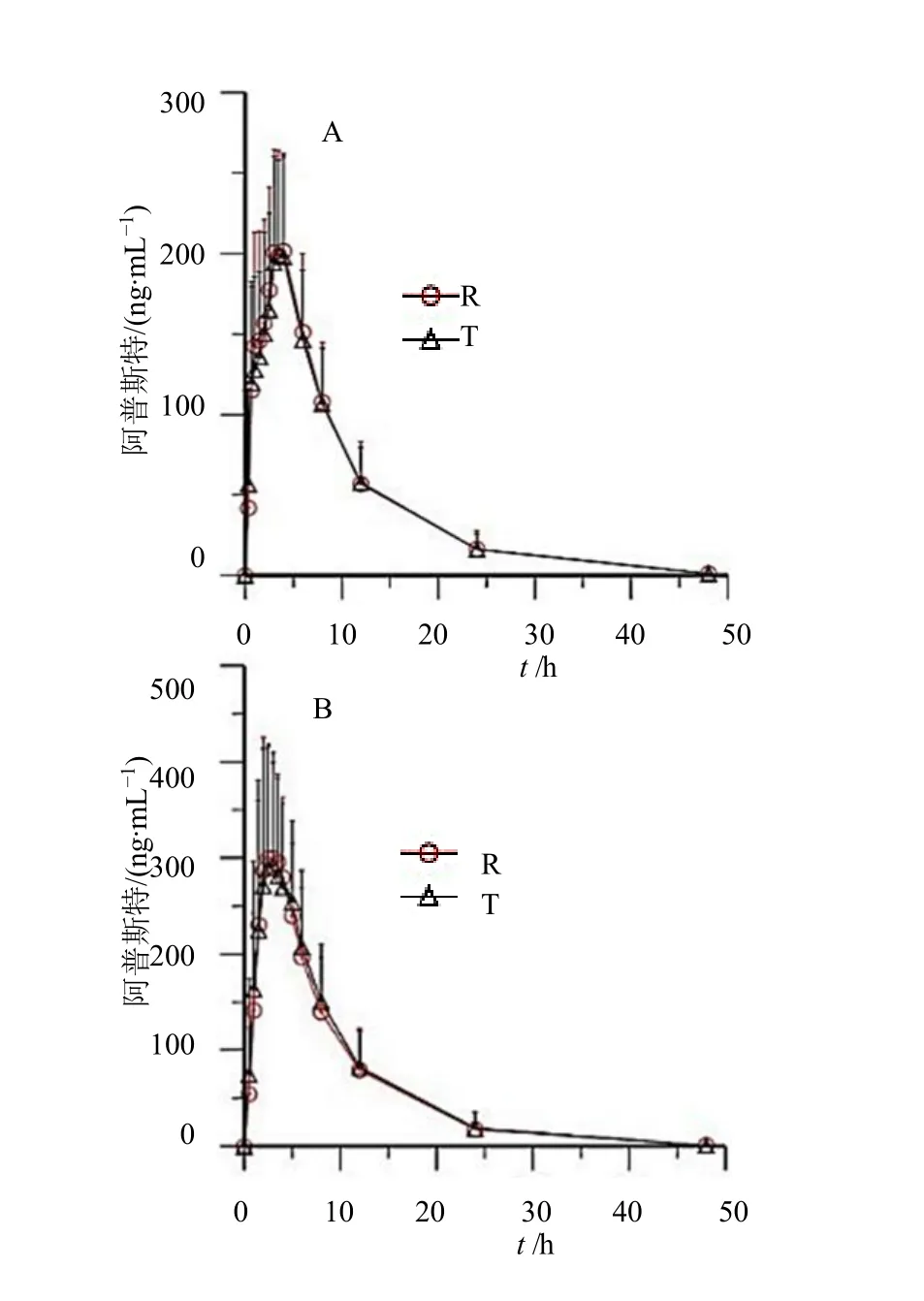
图2 空腹(A)和餐后(B)口服受试制剂和参比制剂后阿普斯特的平均血药浓度-时间曲线Fig.2 Mean blood concentration-time curve of Apremilast Tablets after po administration of test and reference preparation under fasting (A) and fed (B) condition
受试者在空腹和餐后单次口服30 mg 阿普斯特片受试制剂和参比制剂的药动学参数结果见表3。

表3 阿普斯特片受试制剂和参比制剂的药动学参数Table 3 Pharmacokinetic parameters of test and reference preparations of Apremilast Tablets
Cmax、AUC0-t和AUC0-∞在进行对数转换后,采用交叉试验设计的方差分析,结果空腹和餐后阿普斯特的Cmax、AUC0-t和AUC0-∞给药顺序间、给药周期间、制剂间的差异无统计学意义,受试者(顺序)间的差异有统计学意义。
2.4 生物等效性评价
Cmax、AUC0-t和AUC0-∞在进行对数转换后进行双向单侧t检验,计算受试制剂和参比制剂血浆中阿普斯特Cmax、AUC0-t和AUC0-∞的几何均值比的90%置信区间。当受试制剂与参比制剂的Cmax、AUC0-t和AUC0-∞的几何均值比的90%置信区间在80.00%~125.00%等效区间内,即可判定两种制剂的人体生物等效。阿普斯特片的生物等效性结果见表4。空腹试验1 例受试者(K016)由于隐瞒高血压病史,在筛选前两周服用了降血压药物,不确定是否对后续血药浓度是否产生影响,不纳入生物等效性集(BES),但纳入该例受试者第1 周期的数据后进行敏感性分析,结果见表5。受试制剂和参比制剂的tmax进行两配对样本的非参数Wilcoxon 符号秩和检验,结果见表6。
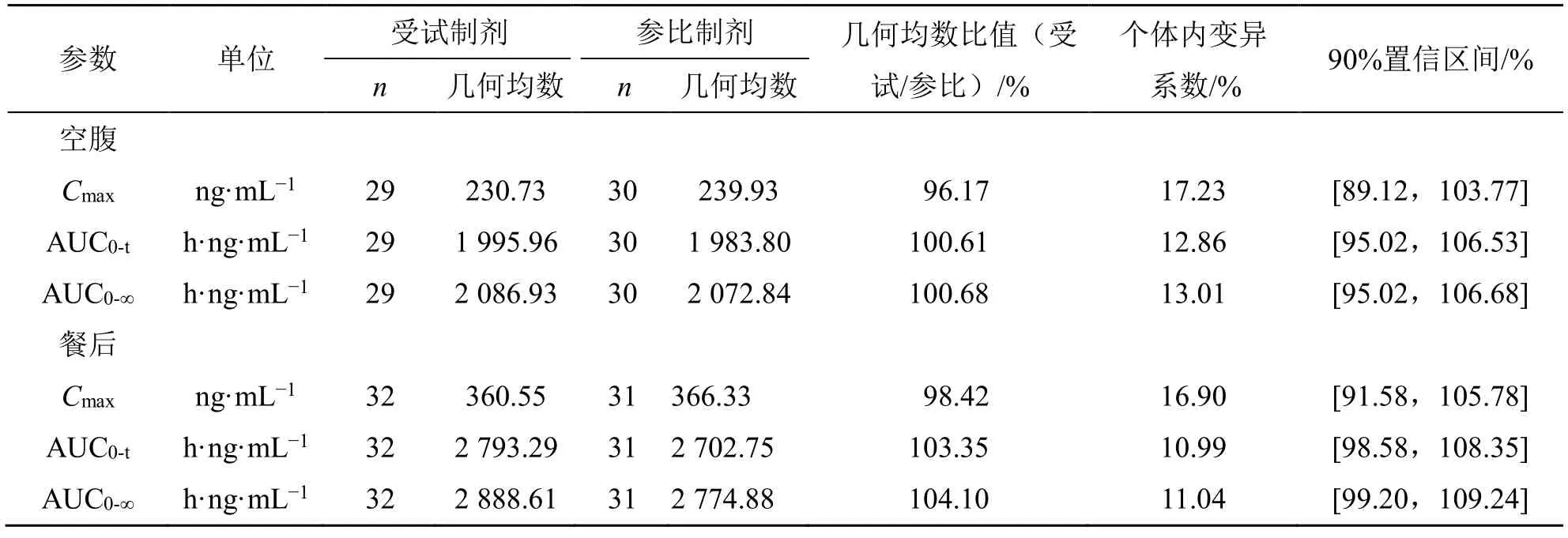
表4 健康受试者空腹和餐后单次口服阿普斯特片后血中阿普斯特的主要药动学参数生物等效性评价(BES)Table 4 Bioequivalence evaluation of main pharmacokinetic parameters in blood of healthy subjects after a single oral administration of Apremilast Tablets under fasting condition and fed condition
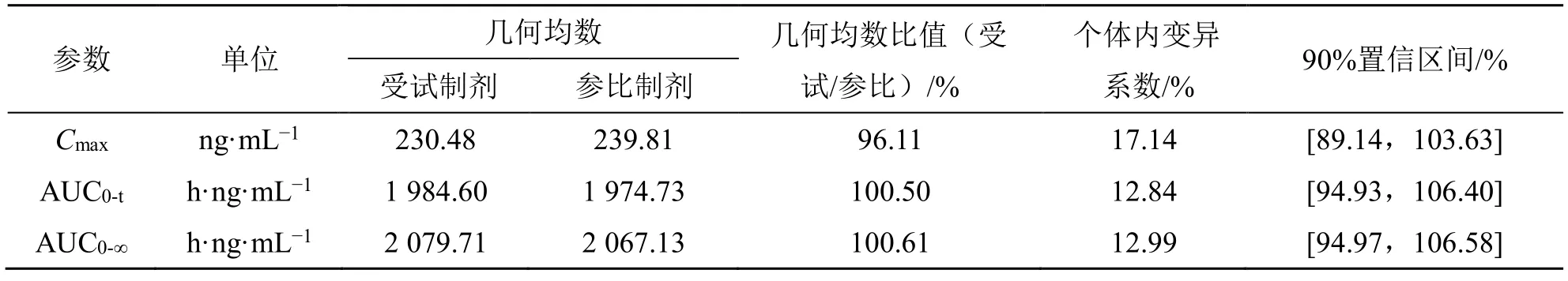
表5 敏感性分析结果Table 5 Sensitivity analysis result

表6 阿普斯特tmax 非参数检验Table 6 Non-parametric test of tmax of apremilast
生物等效性评价结果显示阿普斯特片空腹和餐后试验受试制剂与参比制剂的主要药动学参数Cmax、AUC0-t和AUC0-∞的几何均值比的90%置信区间均落在80.00%~125.00%等效区间内。敏感性分析基于空腹研究中BES,纳入K016 第1 周期数据后,受试制剂和参比制剂的Cmax、AUC0-t、AUC0-∞的几何均值比的 90%置信区间均在 80.00%~125.00%等效区间内,故其等效性评价结果稳定。tmax非参数检验结果显示受试制剂和参比制剂的tmax差异无统计学意义。故认为上述给药顺序的差异可能来源于系统误差,是可以接受的。
综上所述,空腹和餐后条件下阿普斯特片受试制剂和参比制剂在90%的置信区间上Cmax、AUC0-t和AUC0-∞等效,由此支持两种制剂在空腹和餐后条件均具有生物等效性。
3 讨论
阿普斯特片是一种治疗银屑病、白塞病相关性口腔溃疡等的磷酸二酯酶4 小分子抑制剂[2]。参考《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》[10]和《Draft Guidance on Apremilast》[11],采用单中心、随机、开放、两制剂、两序列、两周期交叉设计开展阿普斯特片30 mg 的生物等效性研究,检测分析人血浆中的阿普斯特。
本实验建立了一个灵敏、专属、可靠的用于人血浆中阿普斯特浓度测定的HPLC-MS/MS 分析方法。该分析方法的线性范围为2.00~800 ng/mL。对该分析方法的选择性、基质效应、稳定性、回收率、残留、标准曲线、定量下限等进行了方法验证。结果表明,应用HPLC-MS/MS 法测定人血浆中阿普斯特均符合要求,可用于阿普斯特的试验样品分析。
空腹试验表明健康受试者单次口服阿普斯特片30 mg 受试制剂的血浆药物浓度于给药后3.5 h达到峰值,峰浓度为232 ng/mL。血药浓度达峰后,快速消除,半衰期为6.64 h。结果与参比制剂报道的药动学参数相比[5,8],峰浓度约低31%左右(参比制剂文献报道峰浓度339.86 ng/mL),达峰时间约延长了1 h,推测可能是由于人种或纳入人群(文献中均纳入为男性受试者)导致的差异。餐后试验受试制剂的血浆药物浓度于给药后3 h 达到峰值,峰浓度为371 ng/mL,半衰期为5.32 h。结果与参比制剂报道的药动学参数相比[5,8],峰浓度约高10%左右(参比制剂文献报道的峰浓度为333.85 ng/mL),达峰时间相同,药动学参数基本一致。餐后结果与空腹结果相比,峰浓度约增加30%左右,AUC 增加约29%左右,说明与食物同时给药可能轻微影响阿普斯特的吸收程度,与文献报道[8]的食物影响略有差异,如文献报道阿普斯特平均峰浓度Cmax无增加,且对AUC 的影响不具显著性(增加约10%)。
综上所述,阿普斯特片受试制剂和参比制剂在空腹和餐后状态下单次服用后在健康受试者体内的药动学相似,吸收程度和吸收速度相当,具有生物等效性。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02
中老年保健(2021年3期)2021-08-22
恋爱婚姻家庭(2021年17期)2021-07-27
中国特种设备安全(2021年12期)2021-04-26
今日农业(2020年18期)2020-12-14
中成药(2018年6期)2018-07-11
中成药(2017年4期)2017-05-17
小学生导刊(低年级)(2016年8期)2016-09-24
中国粮油学报(2016年5期)2016-01-23
中国中医药现代远程教育(2014年11期)2014-08-08
