
SMC1A基因变异致Cornelia de Lange综合征2型双胞胎
2023-10-16 07:26张庆华潘海瑞郝胜菊
临床与实验病理学杂志 2023年9期
赵 磊,张庆华,张 钏,陈 雪,潘海瑞,郝胜菊,惠 玲
Cornelia de Lange综合征(Cornelia de Lange syndrome, CdLS)是一种多系统受累的遗传性疾病,临床主要表现为特殊面容、生长发育迟缓、四肢畸形或缺如、先天性心脏病和行为认知障碍。根据表型差异可分为:经典型、温和型和拟CdLS型[1],根据致病基因不同可分为5型,分别与NIPBL、SMC1A、SMC3、RAD21、HDAC8基因相关[2]。SMC1A基因所致的CdLS 2型呈X染色体显性遗传,约占文献报道病例的5%,男女比为1 ∶2[3-4],临床表现较温和,以行为认知障碍为主,可伴有难治性癫痫,面部除眉毛异常外,其它表现轻微,年幼患儿很难依据临床表现明确诊断。本文对2022年3月甘肃省妇幼保健院收治的1例双胞胎患儿的临床资料和家系基因检测结果进行分析,以提高临床医师对该病的认识,利于后期患儿的随访和治疗。
1 材料与方法
1.1 临床资料
1.1.1基本病史 患儿系第3胎第2产(图1、2),胎龄34周,因“先兆早产,双胎”于我院自然分娩,出生体重1 610 g,Apgar评分1 min 9分(呼吸2分、心率2分、皮肤1分、肌张力2分、反应2分),给予清理呼吸道,5 min 10分,脐带绕颈1周,无胎膜早破及胎盘早剥,羊水清亮。患儿出生后因早产伴呼吸急促,三凹征阳性,故以“新生儿呼吸窘迫综合征、低出生体重儿、早产儿”收住新生儿科,分娩前予以地塞米松6 mg/次,共2剂;硫酸镁5 g、15 g、15 g、15 g,共4剂。单绒双羊,患儿弟弟(图3)系第3胎第3产,出生体重1 260 g,与患儿情况一致,同时收住新生儿科。患儿父亲28岁,姐姐7岁,均体健。患儿母亲(图4)乙肝e抗体及核心抗体阳性,妊娠期合并高血压、糖尿病,除眉毛浓密外无明显异常。孕期正常体检,孕12周NT:1.4/1.0 mm,未行唐氏筛查,孕18周四维超声未见明显异常。孕30周超声提示宫内妊娠,双活胎,胎儿如孕27周+4/26周+1,A胎儿双顶径、头尾、腹围、股骨长、肱骨长均低于M-2SD线,羊水偏少;B胎儿均低于M-3SD线。双胎儿脐绕颈1周,体重差异为20%。产前超声提示双胎存活,头位,脐绕颈1周,S/D比值正常,羊水正常低值。
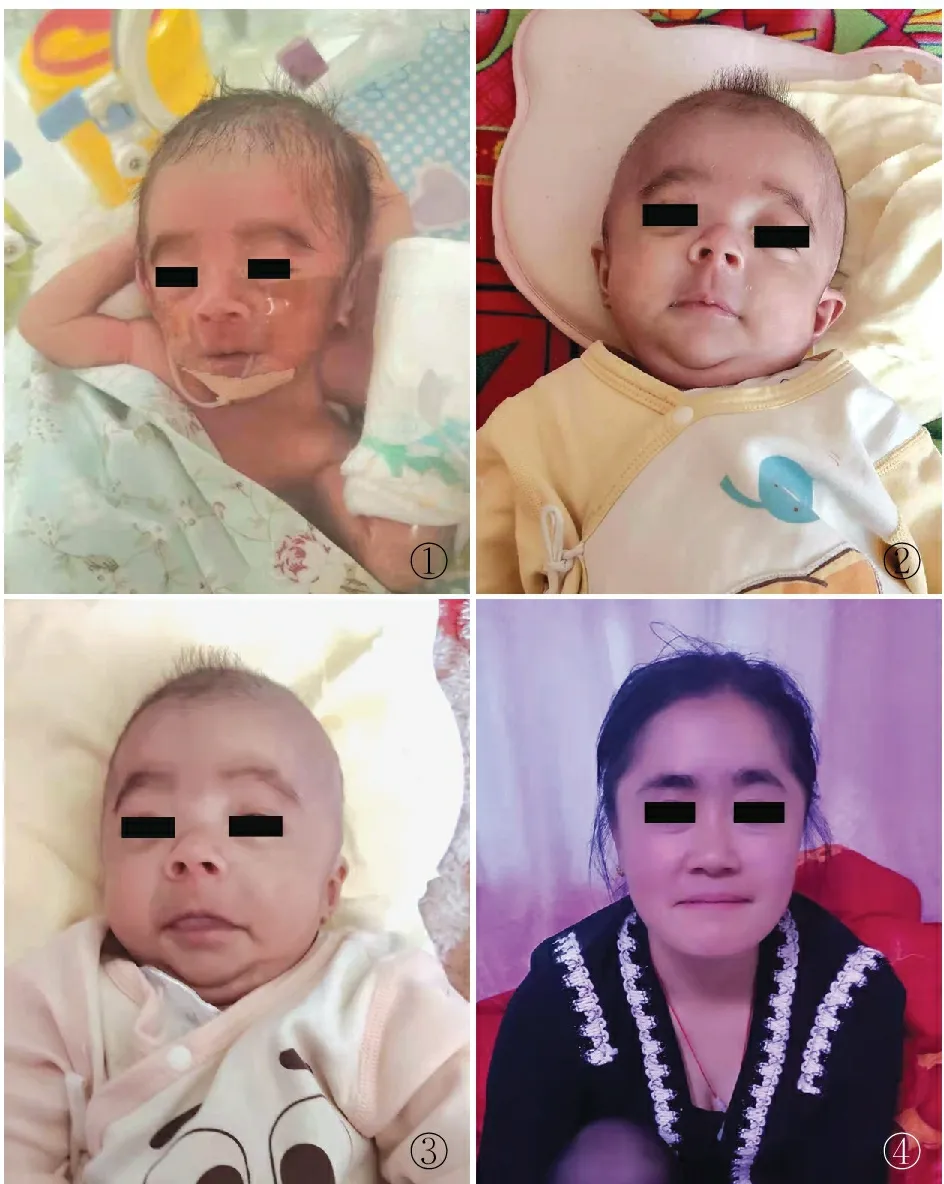
图1 患儿出生时毛发浓密,眉毛呈弓形 图2 患儿3月龄特殊面容 图3 患儿弟弟3月龄特殊面容 图4 患儿母亲眉毛浓密,嘴唇稍薄
1.1.2体格检查 患儿体温36 ℃,脉搏116次/min,呼吸61次/min,SPO2:92%,血压70/36 mmHg,头围28 cm,身长40 cm。特殊面容:眉毛浓密,鼻梁高,鼻孔前倾,嘴唇稍薄,嘴角下斜。神志清楚,反应良好,发育幼稚,呼吸急促,三凹征阳性,腹稍膨隆,肌张力稍弱,关节活动度正常,原始反射未能完全引出。患儿弟弟体温35.8 ℃,脉搏117次/分,呼吸62次/min,SPO2:90%,血压62/37 mmHg,头围27 cm,身长37 cm。

1.1.4治疗与随访 患儿及弟弟入院后完善相关检查,予以CAPA辅助通气、蓝光治疗、静脉输注红细胞、补充磷、粒细胞集落刺激因子等对症治疗。患儿皮肤花纹样改变,腹部膨隆,触之软,肠鸣音弱,腹部平片提示新生儿坏死性小肠结肠炎,予以禁食水、抗感染、静脉高营养等对症支持治疗。经上述治疗后,查体患儿腹软,肠鸣音正常,予以完全水解蛋白奶喂养,患儿纳奶可;2022年5月3日儿童保健科查体一般情况可,营养不良,舌苔白,四肢肌张力高,听力筛查未通过,眼科筛查未做。患儿弟弟精神反应差,皮肤花纹样改变,腹部膨隆,肠鸣音弱,予以禁食水、抗感染治疗,血培养结果回报屎肠球菌阳性,予以头孢呋辛钠抗感染治疗,后腹软肠鸣音正常,予以完全水解蛋白奶喂养,患儿纳奶可;5月16日随访查体:精神反应好,一般情况可,发育良好,皮肤黏膜红润,四肢肌张力正常,脐痂脱落,无渗出,眼底筛查已通过,听力筛查未通过。
1.2 方法
1.2.1标本采集 EDTA抗凝管采集患儿、患儿父母及弟弟外周血各3 mL进行全外显子组测序(北京智因东方诊断公司),本研究经甘肃省妇幼保健院伦理委员会审核通过,患儿父母知情同意。
1.2.2DNA提取 使用血液基因组柱式中量提取试剂盒(康为世纪公司)提取脐血或胎儿组织基因组DNA,操作按照试剂盒说明书进行。所提取DNA样本用Qubit 2.0型荧光计及0.8%琼脂糖凝胶电泳对所提供的DNA样本进行质检,合格后继续后续操作。
1.2.3文库构建 采用IDT公司xGen®Exome Research Panel v2.0捕获探针与gDNA文库序列进行液体杂交,将目标区域DNA片段进行富集,构建全外显子文库,覆盖人基因组中19 396个基因的编码区及部分非编码区,捕获区间大小51 Mb。
1.2.4测序 通过华大智造DNBSEQ-T7测序平台进行高通量测序,目标序列测序覆盖度≥99%,测序过程在智因东方转化医学研究中心完成。
1.2.5生物信息学分析 (1)质控:使用fastp软件对原始数据进行质控,去除接头、低质量reads等;(2)变异检出:使用Burrows-Wheeler Aligner(BWA)软件将测序序列与Ensemble参考基因组GRCh37/hg19比对;应用GATK软件分析出SNP、Indel;最后根据测序深度、变异质量对检测到的SNP、Indel进行过滤和筛选,从而得到高质量可靠的变异;(3)变异注释及预测:使用自主开发的变异注释软件对检测到的高质量变异进行各大数据库(如dbSNP、千人基因组、ExAC、ESP等频率数据库及OMIM、HGMD、ClinVar等)的关联注释。借助Provean、SIFT、Polyphen2-HVAR、Polyphen2-HDIV、M-Cap、Revel、Mutationtaster等蛋白结构预测软件,筛选出对蛋白结构可能有不利影响的变异。
1.2.6Sanger测序验证 利用Sanger测序对阳性位点进行家系验证,引物通过Primer 3.0设计,由兰州天启基因生物公司合成,SMC1A引物序列:正向5′-CATCTCCTTCTC CAGCTCATCC-3′,反向5′-CAAGCTCTCCTTTGGGTGAAAAG-3′,扩增长度249 bp。反应条件:95 ℃预变性5 min;94 ℃ 30 s,60 ℃ 30 s,72 ℃延伸1 min,合计35个循环;72 ℃ 10 min,4 ℃短时保温。
2 结果
通过全外显子组测序分析发现与疾病表型相关的高度可疑变异,Sanger测序确认变异位点并进行了家系验证,结果显示在SMC1A基因7号外显子发现变异位点:SMC1A基因(NM_006306)c.897C>G导致氨基酸改变p.I299M,为错义突变(图5),该变异在不同物种间高度保守(图6)。参照ACMG发布的变异解读指南:错义变异位于致病热点区或CCR区域(图7)(PM1);在正常人群数据库频率<0.000 5(PM2);GnomA D数据库中mis_Z score≥3.09基因上的突变(PP2);两种统计方法(SIFT和REVEL)预测出变异对基因(基因产物)有影响(PP3),该变异初步判定为可能致病,关联疾病为CdLS 2型。

图5 Cornelia de Lange综合征患儿及其父母、弟弟SMC1A基因测序图:患儿、母亲及弟弟均存在SMC1A基因c.897C>G变异位点,父亲为野生型

图6 通过UCSC数据库对变异位点进行保守性分析:该区域序列在不同物种间高度保守

图7 IBS软件[5]绘制SMC1A基因错义变异在SMC1A蛋白结构域上的分布
3 讨论
CdLS又称阿姆斯特丹侏儒症,是一种罕见的遗传性疾病。1933年荷兰儿科医师Cornelia de Lange首次对2例患儿进行了详细描述[6],该综合征在活产儿中的发病率为1/30 000~1/10 000,多为散发病例,少数存在家族发病倾向[7]。尽管全球报道了许多病例,但国内报道病例较少,对于一些表现轻度的患儿,临床医师很难考虑到CdLS,近年来随着分子诊断水平的应用,国内许多病例被发现。与CdLS相关的致病基因主要包括NIPBL、SMC1A、SMC3、RAD21和HDAC8基因,随着对该综合征的研究和认识,一些罕见的基因也被考虑,但由于缺乏典型的临床表现,故临床病例主要以上述5个基因多见,NIPBL基因变异约占70%[7],临床表现较典型。根据CdLS国际共识的诊断标准,依据临床表现也可以明确诊断,但该综合征临床表现存在差异,临床医师对该病认识不足,依据临床表现确诊具有非常大的挑战性,需通过基因检测进一步明确诊断以及CdLS分型。研究发现CdLS相关的致病基因均与黏连蛋白相关,其中SMC1A基因编码黏连蛋白的组成成分,SMC1A蛋白有4个重要的结构域,包括ATP结合位点、卷曲螺旋、Region域和SMC环状结构,在DNA损伤修复、基因转录以及姐妹染色单体分离中起重要作用[8],与赖氨酰氧化酶样蛋白3(lysyl oxidase like 3, LOXL3)结合维持基因组的完整性[9]。检索HGMD数据库发现131个SMC1A基因变异(最后一次访问时间为2022年5月24日),其中剪接变异5例,移码变异17例,缺失变异8例,框内变异11例,错义变异70例,无义变异18例,同义变异2例,通过筛选发现收录81个SMC1A基因变异与CdLS 2型相关。SMC1A基因位于Xp11.22,部分能逃逸X染色体失活,女性患者的致病原因可能是显性负效应,与男性患者相比,女性患者从正常表现到致死性病变均可存在,患儿母亲携带变异位点临床表现却轻微,可能与X染色体失活有关[10]。
本例患儿出生时具有CdLS典型面部特征,基因检测发现与疾病相关的变异位点SMC1A基因c.897C>G,该基因与CdLS 2型相关,临床特征包括面部畸形、手脚畸形、生长延迟、认知障碍、多毛症、胃食管功能障碍和心脏、眼科和泌尿生殖系统异常。虽然目前患儿仅面部特征较典型,但考虑到患儿较小,一些症状尚不明显,对于智力评估、心脏、眼科检查需定期复查。癫痫发作在SMC1A基因变异的个体中约占45%,尤其是截断变异的患儿可表现为难治性癫痫和严重发育不良[7,11],所以对于SMC1A基因变异的患儿,脑电图检测也是必要的。该病临床尚无有效的治疗手段,以对症支持治疗为主,预后主要取决于并发症的严重程度,积极的康复治疗有助于减少并发症,提高患儿生活质量[12]。
总之,本文1例双胞胎患儿SMC1A基因c.897C>G新发变异,患儿及弟弟临床表现基本一致,但实验室检查存在差异,检索数据库和查阅文献发现该变异在国内未见收录和报道,扩大了SMC1A基因变异谱。同时对于表现不明显但存在SMC1A基因变异的女性,在生育指导方面应慎重,尤其当胎儿为男孩时,更应慎重考虑胎儿的去留。目前CdLS的致病机制尚不十分明确,基因型和表型关系复杂,需进一步研究。
猜你喜欢
新民周刊(2022年27期)2022-08-01
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
传染病信息(2021年6期)2021-02-12
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
生物学通报(2019年3期)2019-02-17
百科知识(2015年18期)2015-09-10
生物医学工程学进展(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
