
碳酸羟基磷灰石的生物矿化研究
2010-02-06 12:44朱庆霞徐琼琼罗民华
陶瓷学报 2010年2期
朱庆霞 徐琼琼 罗民华
(景德镇陶瓷学院,景德镇:333001)
1 引言
人体自然骨中磷灰石矿物主要是非化学计量的磷灰石晶体,碳酸根是骨磷灰石中含量最多的掺杂离子。碳酸根能替代羟基磷灰石(HA)晶格中的羟基或磷酸根,分别形成A型或B型替代,或同时占据两个位置,形成AB型替代的碳酸羟基磷灰石(CHA)[1]。在人体骨中的CHA以B型替代为主,A型和B型替代的摩尔比大约为0.7~0.9[2]。对骨科材料在体外生理模拟环境中的生物活性的检测和表征是评价材料性能的一个重要指标。骨生物活性材料在生理环境中可发生一系列离子交换和溶解-沉淀反应,形成生物矿化的碳酸羟基磷灰石(hydroxy-carbonate-apatite, HCA)晶体。HCA在组成和结构上与天然骨的无机矿物类似,可吸附多糖、胶原等细胞外基质,并与宿主骨组织发生骨性结合,促进新骨的形成和生长[3]。多种无机、有机和金属材料都进行过仿生矿化研究[4-6],然而对本身含有碳酸根的CHA材料在生理溶液中的仿生矿化却研究得很少。因此本文将化学组成类骨的CHA和HA浸泡在生理溶液中进行仿生矿化,研究了碳酸根替代对仿生矿化性能的影响,进而对CHA材料的生物活性进行评估。
2 实验
2.1 粉体的制备
HA和CHA粉体的制备:以硝酸钙(Ca(NO3)2· 4H2O)、磷酸氢二铵((NH4)2HPO4)为原料,控制Ca(NO3)2·4H2O与(NH4)2HPO4的摩尔比为1.67,以适当的速度将(NH4)2HPO4溶液滴加到Ca(NO3)2·4H2O溶液中制备HA。以Ca(NO3)2·4H2O、(NH4)2HPO4和NaHCO3为原料,控制Ca(NO3)2·4H2O,(NH4)2HPO4和NaHCO3的摩尔比,以适当的速度将 (NH4)2HPO4和NaHCO3的混合溶液滴加到Ca(NO3)2·4H2O溶液中制备CHA。在整个反应过程中,通过滴加氨水调节溶液的pH值在10~11之间,反应温度为90℃。反应结束后,继续搅拌3 h,然后在室温下陈化24 h,再用蒸馏水洗涤、离心、过滤、120℃干燥。
2.2 样品的制备
将CHA和HA粉体在250 MPa等静压作用下压成块体,将CHA块体置于自制的通有湿二氧化碳气体(其中湿气的带入是将气体在进入石英管式炉前,流经室温下盛有蒸馏水的三口瓶而实现的)的石英管式炉内进行热处理。作为对比,将HA块体在空气气氛中加热处理。控制两组样品经Archimedes法测试的致密度均为91%,其中CHA样品经FTIR和元素分析,碳酸根含量为5.72wt%,A型和B型替代的摩尔比为0.72。两组样品均经过No.1000防水砂纸抛光处理,然后在双蒸馏水中超声清洗。

表1 人体血浆和模拟生理溶液(S B F)的各种无机离子浓度(10-3mo l/L)Tab.1 Ion concentration of human plasma and SBF solution(10-3mol/L)

图1 C H A在S B F溶液中反应不同时间的S E M形貌Fig.1 SEM morphologies of CHA reacted with SBF for different times(a)6 days;(b)12 days;(c)24 days
2.3 模拟体液的配制
配制SBF溶液所使用的化学药品均为分析纯化学试剂。溶液在37℃水浴恒温条件下进行配制,各药品在磁力搅拌下按顺序缓慢加入去离子水中,溶液始终为清澈透明状态。一旦出现轻度乳浊,则说明有沉淀物形成,溶液则不能再用。最终溶液用HEPES(N-(2-羟乙基)-对二氮己环N'-(4-丁烷磺酸))调整到pH=7.4,密封于玻璃容器中并置于冰箱内冷藏保存。模拟生理溶液的组成如表1所示。作为对照,表中同时列出了人血浆中的各种无机离子浓度。
2.4 样品性能的表征
用 X射线衍射(X-ray diffraction,XRD)仪(PANalytical X'pert PRO,Netherlands)慢速扫描确定物相,工作条件为管压40 kV,管流40 mA,2θ的步长为0.02°,实验结果与JCPDS卡片对照分析。用Fourier变换红外(Fourier transform infrared,FTIR)光谱仪(Vector 33,Germany)分析样品的分子基团,特别是碳酸根的振动。用躺滴法(A sessile drop method)接触角测定仪(OCA15,dataphysics,Germany)测量实验液体在样品表面的接触角以计算表面能。采用LECO1530VP型扫描电子显微镜进行表面形貌观察,并用日本日立公司生产的Z-5000型原子吸收光谱仪分析了SBF溶液与材料反应不同时间后,钙离子浓度的变化。

图2 H A在S B F溶液中反应不同时间的S E M形貌Fig.2 SEM morphologies of HA reacted with SBF for different times(a)12 days;(b)18 days;(c)24 days
3 结果与讨论
对CHA和HA两组样品进行仿生矿化,每组平行试样不少于5个。分别将各组样品浸入盛有SBF的密闭聚乙烯瓶中,其中SBF溶液的体积按试样表面积与溶液的体积比为0.1cm-1标准量取。保持37.0℃恒温,对不同反应时间的SBF溶液分别取样,置于冰箱中冷藏保存,用于溶液中离子浓度的分析。每组试样于不同反应时间后取出,用去离子水冲洗,室温80℃烘箱干燥,用于矿化涂层的成分分析和形貌分析。
3.1 矿化层表面形貌分析
在SBF中浸泡不同的时间,取出后进行SEM观察,图1和图2分别为CHA和HA经不同矿化时间的样品表面形貌。
CHA矿化6天后,可见低结晶度的HCA晶核沉析在样品表面,大小约为30~40nm,在表面的分布较为均匀(图1a);反应12天后,材料表层被长度约为150~200nm的叶片状HCA微晶层所覆盖(图1b),随着反应时间的延长,HCA层继续生长,反应24天形成HCA球形晶簇,其直径在200~300nm,球形晶簇相互融合,形成板状HCA覆盖层(图1(c))。

图3 样品动态接触角Fig.3 Dynamic contact angles between(a)CHA and propane trio;(b)HA and propane trio;(c)CHA and SBF;(d)HA and SBF
由图2所示,HA样品的矿化过程与CHA类似,但是矿化层在HA样品上的沉积速度要低于CHA样品。
在SBF溶液中生物活性材料表面形成HCA晶核是非均匀成核过程[7-8],CHA中由于碳酸根的替代,致使晶体结构畸变,溶解度增加[9-10],CHA样品表面Ca2+、PO43-离子的溶出量大于HA样品表面的溶出量,一方面使得溶液中Ca2+、PO43-浓度增大,过饱和度增加,而成核速率对过饱和度的变化非常敏感,因此,表面微区内Ca2+、PO43-离子过饱和度的增加必将提高样品表面晶核的形成速率;另一方面材料表面钙磷化合物的溶出,使得表面缺陷增多,增加了表面成核位点,也有利于增加晶核的形成量。
同时,表面能的影响不可忽视,表面能越大,在形成矿化层的过程中,吸附和反应更容易在高能表面上发生。图3分别为生理溶液和丙三醇在样品CHA和HA表面形成的动态接触角。根据接触角的测试数据,表面能采用Owens-Wendt-Kaeble's方程[11]计算求得CHA的表面能为50±2mJ/m2,而HA的表面能为43±2 mJ/m2。固体表面的暴露基团和固体表面粗糙度等对接触角有明显的影响[12]。CHA和HA在测试前经过相同的样品制备过程,其表面粗糙度和致密度基本一致,故主要是碳酸根的替代导致样品表面组分发生变化,从而进一步影响表面能。S.A.Redey[13-14]等人发现A型替代CHA的表面能低于HA的表面能,从而导致破骨细胞在其表面吸附量较少,胶原的生成量也较少。而本实验中CHA的替代类型是以B型为主,这种类型的替代导致了表面能升高,磷灰石晶体在其表面形核时接触角小,所需克服的形成能就较小,核化速率大,形成矿化层的时间短,这与SEM结果吻合。

图4 S B F浸泡液中C a2+浓度随浸泡时间的变化曲线Fig.4 The change of Ca2+concentration in SBF solution with reacting times
当样品在模拟体液中浸泡时,Ca2+的溶解与消耗使得样品表面与模拟体液间形成Ca2+的浓度梯度层。图4所示为模拟体液中Ca2+浓度随浸泡时间变化的曲线图。在浸泡的最初阶段,由于样品表面Ca2+的溶解速度大于其沉积速度,因此浓度梯度层中靠近样品表面部分的Ca2+浓度大于靠近模拟体液部分的Ca2+浓度,Ca2+逐渐由样品表面向模拟体液中扩散。Ca2+浓度明显上升。CHA样品浸泡6天后,溶液中Ca2+浓度达到最大值,约132ppm,而HA样品溶解速度较慢,并且溶解度较低,在浸泡12天后,溶液中Ca2+浓度才达到最大值,约117ppm。此后由于HCA在样品表面析出,消耗了溶液中的Ca2+,溶液中Ca2+浓度逐渐减少,并趋于稳定。
3.2 矿化层的分析
通过衰减全反射红外光谱对浸泡后的试样表面进行测试。图5为HA样品在SBF浸泡不同时间后的FTIR谱图。所有样品都出现羟基的摆动振动峰(630cm-1,ν2)、磷酸根的非对称伸缩振动峰 (1090~1044cm-1,ν3)和对称伸缩振动峰(960cm-1,ν1)。浸泡前和浸泡6天后的样品表面并没有明显的碳酸根的振动峰,表明在这个过程中并没有形成具有HCA结构的矿化层。当浸泡12天后,谱图中开始出现CO32-非对称伸缩振动吸收带(1400~1500 cm-1,ν3)和弯曲振动吸收峰(870~880cm-1,ν2),并且ν3分裂成双峰,这是HA中CO32-基团的特殊振动模式[15]。表明随着浸泡时间的延长,通过HA样品表面的溶解,SBF溶液的过饱和度逐渐增加,形成的磷灰石晶核便会通过消耗溶液中的Ca2+、PO43-、CO32-等离子自发长大,从而形成在组成和结构上都与骨组织类似的HCA矿化层。
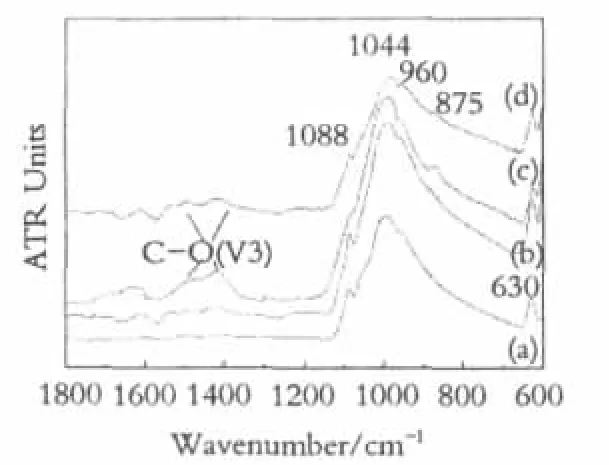
图5 H A样品在S B F溶液中反应不同时间的F T I R谱图Fig.5 FTIR spectra of HA sample reacted with SBF for different times(a)before soaking;(b)6 days;(c)12 days;(d)24 days
图6为CHA样品在SBF浸泡不同时间后的FTIR谱图。它同样具有典型的磷灰石类矿物红外光谱的特点。但与HA样品组不同的是,CHA样品组碳酸根的振动峰较强,碳酸根含量较高。这是由于CHA含有较高的碳酸根含量,溶解度高,能溶解较多的碳酸根到SBF溶液中,故在形成矿化层时,可以有更多的碳酸根掺杂到磷灰石结构中。红外光谱中特征吸收带的宽窄、尖锐程度以及是否发生间并分裂,可以反映矿物的结晶程度[16]。在图6a中可以看出CHA样品中ν3-CO3尖锐的双峰和明显的ν3-PO4和ν1-PO4峰。在图6 (b),(c)中,CO32-的ν3振动吸收带加宽,以致两峰相连而合并,并且1088 cm-1处和960cm-1处PO43-峰消失,只在1100~900 cm-1处出现一个宽而钝的弥散峰,表明此时形成的矿化层结晶度比较低。但当浸泡时间延长为24天,ν3-CO3重又发生分裂,ν1-PO4变得比较清晰,出现了较弱的1088cm-1处PO43-峰,吸收峰发生分裂且峰形尖锐,说明在反应的后期,矿化层由无定形态向晶态过渡,结晶程度不断提高。
3.3 矿化层的分析
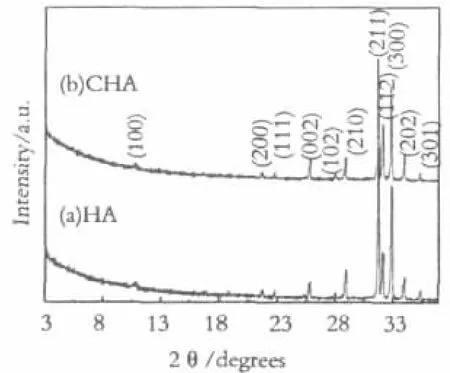
图7 样品表面矿化层的X R D谱图Fig.7 XRD spectra of the samples with mineralized layers
图7给出了HA和CHA样品在SBF中矿化12天后表面XRD谱图。两种样品矿化层的主晶相均为HA。Xiong[17]等经过计算,认为在pH=7.4的生理溶液环境中,磷酸八钙(OCP)的成核速率是大于CHA和HA的,故沉积层晶相往往是由OCP和HA组成的。由于OCP和HA的衍射峰在高角度处大部分互相重合而不易区别,故进行了样品的小角衍射,但在谱图中并未出现位于2θ=4.72°处的OCP(010)晶面的特征衍射峰。这是由于在生理条件下,OCP是热力学亚稳相,浸泡12天后,OCP已经完全转化为热力学稳定的HA相,无其他晶相存在。
4 结论
利用体外仿生矿化的方法评估了碳酸羟基磷灰石的生物活性。研究结果表明:
(1)碳酸羟基磷灰石样品在模拟生理溶液中可形成表面类似天然骨中矿物的碳酸羟基磷灰石层(HCA)。
(2)B型替代为主的CHA样品矿化6天后,可见低结晶度的HCA晶核沉析在样品表面;随着浸泡时间的延长,生成的晶体是叶片状HCA,最后形成相互融合的球形晶簇,覆盖于整个样品表面。
(3)B型替代为主的CHA材料较高的溶解度和表面能导致矿化层形成速度较快,与HA样品相比,CHA矿化能力较好,具有较高的生物活性。
1李世普.生物医用材料导论.武汉:武汉工业大学出版社,2000
2 Rey C,Collins B,Goehl T,et al.The carbonate environment in bone mineral:a resolution-enhanced Fourier transform infrared spectroscopy study.Calcif.Tissue Int,,1989,45(3):157~164
3 Ducheyne P and Qiu Q.Bioactive ceramics:the effect of surface reactivityonboneformation and bone cell function.Biomaterials, 1999,20:2287~2303
4 Ni J.and Wang M.Materials Science and Engineering:C,2002, 20:101~109
5 Roether J A,Boccaccini A R,Hench L L,et al.Development and in vitro characterisation of novel bioresorbable and bioactive composite materials based on polylactide foams and bioglass(R)for tissue engineering applications.Biomaterials, 2002,23:3871~3878
6 Chu T M G,Orton D G,Hollister S J,et al.Mechanical and in vivo performance of hydroxyapatite implants with controlled architectures.Biomaterials,2002,23:1283~1293
7杨华明,宋晓岚,金胜明.新型无机材料.北京:化学工业出版社, 2005
8徐恒钧.材料科学基础.北京:北京工业大学出版社,2001
9 Tang R,Henneman Z J and Nan collas G H.Constant composition kinetics study of carbonated apatite dissolution. Journal of Crystal Growth.2003,249:614~624
10 Ito A,Maekawa K and Tsutsumi S.Solubility product of OH-carbonated hydroxyapatite.J.Biomed.Mater.Res.,1997, 36:522~528
11 Kokubo T,Kim H M and Kawashita M.Bone Engineering. Canada,2000:190~194
12胡纪华,杨兆禧,郑忠.胶体与界面化学.广州:华南理工大学出版社,1999:187~189
13 Redey S A,Razzouk S,Rey C,Bernache-Assolant D,Leroy G,Nardin M and Cournot G.Osteoclast adhesion and activity on synthetic hydroxyapatite,carbonated hydroxyapatite,and natural calcium carbonate:Relationship to surface energies.J. Biomed.Mater.Res.,1999,45:140~147
14 Redey S A,Nardin M,Bernache-Assolant D,et al.Behavior of human osteoblastic cells on stoichiometric hydroxyapatite and type A carbonate apatite:Role of surface energy.J.Biomed.Mater.Res.,2000,50:353~364
15 Vignoles M,Bonel G and Holco mb D W.Influence of preparation conditionson the composition oftype B carbonated hydroxyapatite and on the localization of the carbonate ions.Calcif.Tissue Int.,1988,43(2):33~40
16朱庆霞.碳酸羟基磷灰石粉体和涂层的制备、结构及性能研究.广州:华南理工大学,2007
17 Xiong L and Yang L.T heoreticalanalysis of calciu m phosphate precipitation in simulated body fluid.Biomaterials, 2005,26:1097~1108
猜你喜欢
能源工程(2022年1期)2022-03-29
小天使·二年级语数英综合(2021年5期)2021-07-11
昆明医科大学学报(2021年1期)2021-02-07
湿法冶金(2019年5期)2019-10-18
云南医药(2019年3期)2019-07-25
中学化学(2017年2期)2017-04-01
现代工业经济和信息化(2016年3期)2016-05-17
湖北科技学院学报(医学版)(2015年3期)2015-02-28
无机化学学报(2014年7期)2014-02-28
中国洗涤用品工业(2011年4期)2011-03-20
