
MgB 和MgB2(1A1)的结构与解析势能函数*
2010-09-08 06:05韩晓琴蒋利娟刘玉芳
物理学报 2010年7期
韩晓琴蒋利娟刘玉芳
1)(河南师范大学物理与信息工程学院,新乡453007)
2)(商丘师范学院物理与信息工程系,商丘476000)
3)(新乡学院物理系,新乡453003)
(2009年7月9日收到;2009年9月28日收到修改稿)
MgB 和MgB2(1A1)的结构与解析势能函数*
韩晓琴1)2)蒋利娟3)刘玉芳1)†
1)(河南师范大学物理与信息工程学院,新乡453007)
2)(商丘师范学院物理与信息工程系,商丘476000)
3)(新乡学院物理系,新乡453003)
(2009年7月9日收到;2009年9月28日收到修改稿)
分别采用QCISD/6-311G和QCISD/6-311++G(df)方法,对MgB和MgB2分子的微观结构进行理论计算.在此计算基础上,运用多体展式理论方法,推导出MgB2分子的解析势能函数,其等值势能面图准确再现了MgB2分子的结构特征及势阱深度,并讨论了B+MgB和Mg+BB分子反应的势能面特征.这些结果可用于微观反应动力学的研究.
MgB,MgB2,分子结构,势能函数
PACC:3120,3130,3520D,3520G
1. 引言
MgB2作为新型的超导材料已取得了很多研究成果[1—5].在已发现的常规超导材料中,MgB2的中界转变温度最高且化学性质稳定,因此在应用方面具有很大的潜力.2001年3月初,日本科学家Nagamatsu等[6]发现MgB2在39 K左右时表现出超导性.2004年,韩克利等[7]采用QCISD/6-311G*和CCSD(T)/CC-PVTZ两种方法对MgB2分子的稳态结构和振动特性进行了研究;2006年,陈玉红等[8]研究了MgB2团簇结构与性质的密度泛函理论.同年,美国宾州大学的李奇等[9]仔细测量了剩余电阻极小的MgB2薄膜的磁阻.为了对MgB2分子有更多的了解,本文在Gaussian03程序下,采用QCISD/6-311++G(df)方法对MgB2分子的平衡几何、离解能、谐振频率和力常数等进行了计算,利用多体项展式理论推导出MgB2分子基态的势能函数,并根据势能函数讨论了B+MgB,Mg+B2的反应机理.为进一步研究动力学特性提供了理论依据.
2. 基态MgB2的电子状态与离解极限
为了准确表达体系的势能函数,须确定在分子离解极限中各种原子和原子团的电子状态.若Mg和B原子分别为基态1Sg和2Pu,则组合1Sg+2Pu只能得到二重态,而由从头计算结果[8]给出MgB (X4Σ-)原子团的基态为四重态,因此,Mg和B原子不能同时为基态电子态.
若B原子为基态2Pu,则组合2Pu+4Σ-只能得到三重与五重态,而文献[7,8]表明MgB2体系基态为单重态.若B原子为激发态4Pg[10],则基态MgB和激发态B生成MgB2分子的电子状态可表述为

同理,若Mg为基态1Sg,则Mg(1Sg)和B2(X3Σg-)[11]只能得到三重态,若Mg为激发态3Pu,则生成MgB2的电子状态可表述为
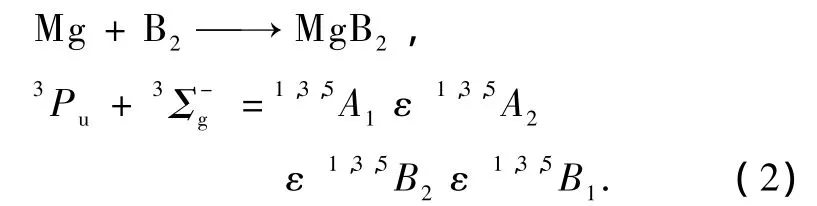
从头计算结果给出的电子状态为1A1,与(1)和(2)式对比,得出MgB2的基态为1A1(C2V群).根据微观过程的可逆性原理[12],可得基态MgB2的可能离解极限为

由上述离解极限可求得基态MgB2分子的离解能为
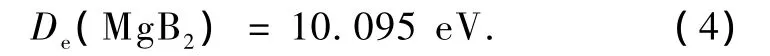
3. MgB2分子的平衡结构及其力学性质
结构与性质是多体项展式分析势能函数的基础.本文采用Gaussian03程序,用QCISD方法,在6-311++G(df)基组水平上,对MgB2分子的键长R、键角αBMgB、离解能De、力常数f、谐振频率ω(对称伸缩振动频率ω1(a1)、弯曲振动频率ω2(b2)、反对称伸缩振动频率ω3(a1))等进行了计算,结果见表1.

表1 MgB2分子的结构与性质常数
从结构优化可知,MgB2具有C2V构型,其基态为1A1.
4. MgB,B2的结构、性质及解析势能函数
本文采用QCISD/6-311G方法,对MgB分子进行了几何优化,在优化的基础上对分子进行频率计算,并对基态进行单点能扫描.结果表明基态为4Σ-,平衡核间距为0.2161nm,谐振频率为533.6611cm-1,De为2.261 eV.通过理论计算得到的结果与实验值和文献值符合得非常好.对得到的一系列单点势能值采用最小二乘法,拟合势能函数[13]为

其中ρ=R-Re,R,Re分别为双原子分子的核间距和平衡间距,De为离解能,a1,a2,a3为拟合参数,其结果列于表2.
图1给出了MgB(4Σ-)的单点能量值和拟合势能曲线.可以看出在计算范围内扫描得到的能量点与拟合势能函数重合,正确表达了MgB分子的势能随核间距的变化趋势.由Murrell-Sorbie势能函数参数与力常数的关系以及力常数f2,f3,f4与光谱数据的关系[13],求得分子的光谱数据和力常数.结果见表3.

表2 B2[13]和MgB的Murrell-Sorbie势能函数参数

表3 基态B2和MgB的力常数与光谱常数
5. 基态MgB2的多体项展式势能函数
对于MgB2分子,设基态原子能量为零,由多体项展式理论方法,得到MgB2分子的势能函数为

其中R1,R2为Mg和B两原子之间的核间距(R1= R2=RMgB),R3为两个B原子之间的核间距(R3= RBB).(6)式中V(2)MgB(R1),V(2)MgB(R2),V(2)BB(R3)为两体项MgB(X4Σ-),B2(X3Σ-g)的势能函数,势能函数参数见表2.V(3)MgB2(R1,R2,R3)为三体项MgB2(X1A1)的势能函数,通常可表示为多项式P和量程函数T的乘积[13]

式中P,T分别表示为
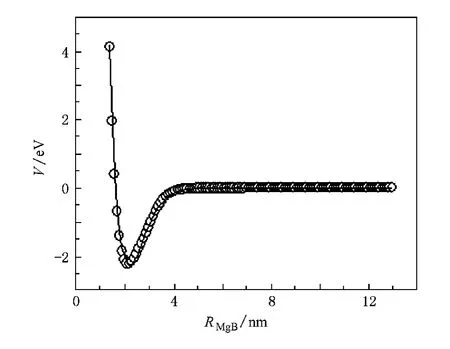
图1 MgB(4Σ-)的分子势能曲线
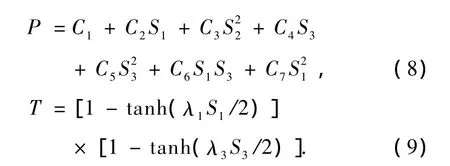
基态MgB2分子为C2V构型,为了方便研究势能函数,根据势能面的结构特征,采用优化内坐标.取MgB2的两个平衡键长为参考结构,R01=R02=RMgB=0.222nm,R03=RBB=0.15673nm.故内坐标ρi经下列变换成为优化内坐标Si,形式为
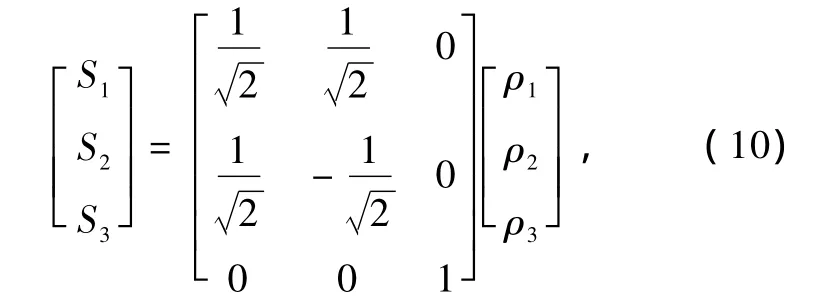
其中ρi=Ri-RiS2对R1和R2的交换是反对称的(i =1,2,3),但R1和R2交换后分子是相同的,为了满足这一物理意义,S2只能含偶次项.(8)和(9)式中分别有7个线性系数和两个非线性系数λ1,λ3.对势能面进行非线性优化,可以确定出两个非线性系数λ1,λ3,而7个线性系数由7个线性方程组求解得出.结果列于表4中,势能函数(6)式的等值势能图如图2—4所示[14—19]:
图2是固定αBMgB=41.34°时,对势能函数(6)式绘制的对称伸缩振动等值势能面图,图中正确再现了MgB2分子的C2V结构特征.在平衡点RMgB= 0.222nm处,有一势阱,其深度为10.095 eV,而且在B+MgB→MgB2反应中没有鞍点存在,不存在明显的势垒,容易生成B—Mg—B络合物,是一个很容易进行的无阈能反应.0
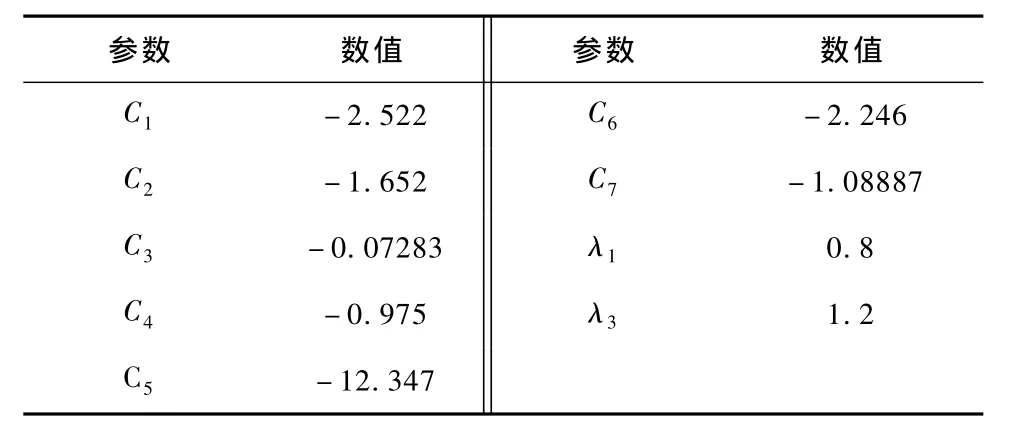
表4 MgB2势能函数的三体项参数

图2 MgB2(1A1)分子伸缩振动的等值势能图(等值线的间隔为0.5 eV)
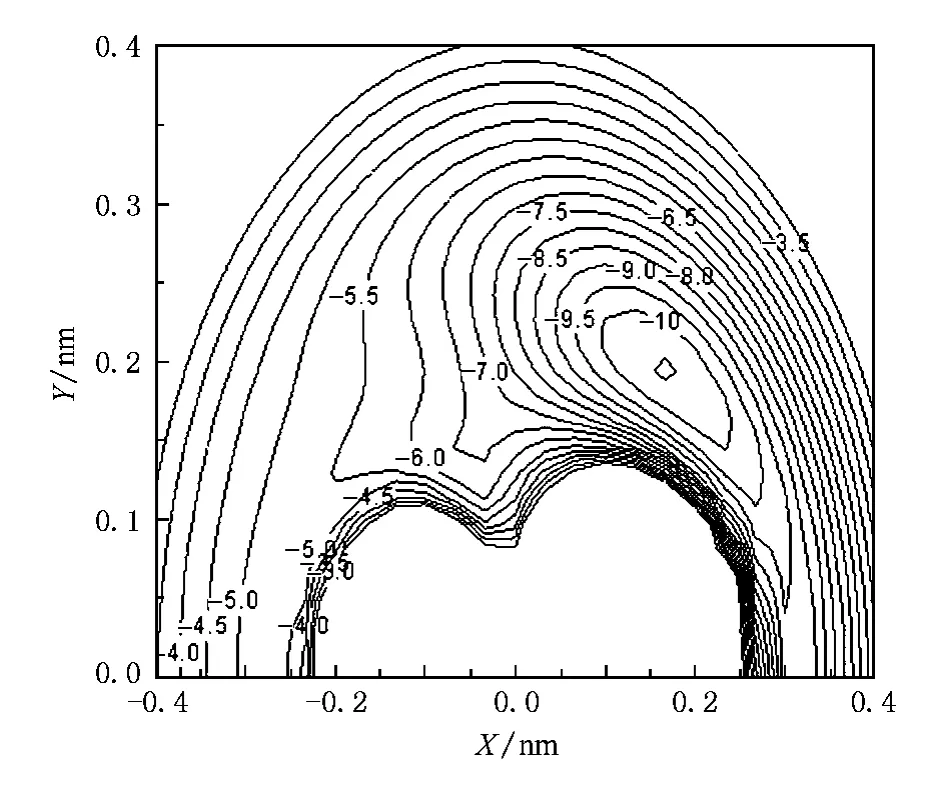
图3 B绕Mg—B(RMgB=0.222nm)旋转的等值势能图(等值线的间隔为0.5 eV)
图3和图4分别为固定Mg—B键和B—B键在X轴上的旋转等值势能图.它们都能清晰地表明,在αBMgB=41.34°,RMgB=0.222nm处,出现一势阱,其深度为10.095 eV.从图3可以看出,B原子从与Mg—B键垂直的方向进攻Mg—B时生成较稳定的MgB2分子.图4体现了基态MgB2的C2V对称性,同时表明Mg原子从与B—B键垂直的方向进攻B—B生成较稳定的MgB2分子.

图4 Mg绕B—B(RBB=0.15673nm)旋转的等值势能图(等值线的间隔为0.5 eV)
6. 结论
本文采用QCISD/6-311++G(df)方法,对MgB2(1A1)分子进行了优化计算,得出它的电子状态为X1A1,MgB2分子具有C2V对称性,其αBMgB= 41.34°,平衡核间距RMgB=0.222nm,离解能De= 10.095 eV,并计算出谐振频率ω1(a1)=436.817cm-1,ω2(b2)=471.585cm-1,ω3(a1)=1115.728cm-1.其理论结果与实验值符合很好.采用QCISD/ 6-311G方法,对MgB(4Σ-)分子进行了优化计算,得到了它的微观几何结构、力学性质和光谱数据.用最小二乘法拟合出MgB分子的Murrell-Sorbie势能函数.在此基础上,采用多体项展式方法导出了基态MgB2分子的解析势能函数及三种等值势能图,其等值势能图从不同角度检验了势能面是否符合三原子分子的几何构型,正确复现了MgB2的平衡结构特征和能量变化信息,并给出一些动力学信息.
[1]Bud’ko S L,Lapertot G,Petrovic C,Cunningham C E 2001 Phys.Rev.Lett.86 1877
[2]Hirsch J E,Marsiglio F 2001 Phys.Rev.B 64 144523
[3]Kortus J,Mazin I I,Belashchenko K D 2001 Phys.Rev.Lett. 86 4656
[4]Lorenz B,Meng R L,Chu C W 2001 Phys.Rev.B 64 12507
[5]Schmidt H,Zasadzinski J F,Gray K E,Hinks D G 2001 Phys. Rev.B 63 220504
[6]Nagamatsu J,Nakagawa N 2001 Nature 410 63
[7]Yang C L,Zhang X,Han K L 2004 J.Mol.Stru.(Theochem) 677 11
[8]Chen Y H,Zhang C R,Ma J 2006 Acta Phys.Sin.55 171(in Chinese)[陈玉红、张材荣、马军2006物理学报55 171]
[9]Li Q,Liu B T,Hu Y F,Chen J 2006 Phys.Rev.Lett.96 167003
[10]Liu Y F,Han X Q,Lü G S,Sun J F 2007 Acta Phys.Sin.56 4412(in Chinese)[刘玉芳、韩晓琴、吕广申、孙金锋2007物理学报56 4412]
[11]Dupuis M,Liu J 1978 Chem.Phys.68 2902
[12]Zhu Z H 1996 Atomic and Molecular Reaction Statics(Beijing: Science Press)(in Chinese)[朱正和1996原子与分子反应静力学(北京:科学出版社)]
[13]Zhu Z H,Yu H G 1997 Molecular Structure and Potentical Energy Function(Beijing:Science Press)(in Chinese)[朱正和、俞华根1997分子结构与势能函数(北京:科学出版社)]
[14]Ni Y,Jiang G,Zhu Z H 2004 Acta Phys.Chim.Sin.20 1380 (in Chinese)[倪羽、蒋刚、朱正和2004物理化学学报20 1380]
[15]Hisatomo H 2002 Physica C 378 18
[16]Sun J F,Wang J M,Shi D H,Zhang J C 2006 Acta Phys.Sin. 55 4490(in Chinese)[孙金锋、王杰敏、施德恒、张计才2006物理学报55 4490]
[17]Zhu Y,Jiang G,Fang F,Zhu Z H 2006 Acta Phys.Chim.Sin. 22 538(in Chinese)[朱瑜、蒋刚、方芳、朱正和2006物理化学学报22 538]
[18]Jiang L J,Liu Y F,Liu Z Z,Han X Q 2009 Acta Phys.Sin.58 0201(in Chinese)[蒋利娟、刘玉芳、刘振中、韩晓琴2009物理学报58 0201]
[19]Zhao J,Cheng X L,Yang X D,Zhu Z H 2009 Acta Phys.Sin. 58 5280(in Chinese)[赵俊、程新路、杨向东、朱正和2009物理学报58 5280]
PACC:3120,3130,3520D,3520G
*Project supported by the National Natural Science Foundation of China(Grant No.10574039),the Key Program of Science and Technology Research Foundation of Ministry of Education of China(Grant No.206084),and the Innovation Talents Program of Institution of Higher Education of Henan Province,China(Grant No.2006KYCX002).
†Corresponding author.E-mail:yf-liu@henannu.edu.cn
Structure and potential energy function of MgB and MgB2(1A1)*
Han Xiao-Qin1)2)Jiang Li-Juan3)Liu Yu-Fang1)†
1)(College of Physics and Information Engineering,Henan Normal University,Xinxiang453007,China)
2)(Department of Physics and Information Engineering,Shangqiu Normal University,Shangqiu476000,China)
3)(Department of Physics,Xinxiang University,Xinxiang453003,China)
(Received 9 July 2009;revised manuscript received 28 September 2009)
Quadratic configuration interaction(QCISD)method has been used to optimize the possible ground state structures of MgB and MgB2by the 6-311G and 6-311++G(df)basis sets.The potential energy functions of MgB2have been derived from the many-body expansion theory.The potential energy functions describe correctly the configurations and the dissociation energies of the two ground-state molecules.Molecular reaction kinetics of B+MgB and Mg+BB based on the potential energy functions is discussed briefly,which is successfully used for describing molecular reaction dynamics.
MgB,MgB2,molecular structure,potential energy function
book=378,ebook=378
*国家自然科学基金(批准号:10574039)、教育部科学技术研究重点项目基金(批准号:206084)和河南省高校杰出科研人才创新工程(批准号:2006KYCX002)资助的课题.
†通讯联系人.E-mail:yf-liu@henannu.edu.cn
猜你喜欢
——《势能》
文化纵横(2022年3期)2022-09-07
中学生数理化·八年级物理人教版(2022年6期)2022-06-05
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
中学生数理化·八年级物理人教版(2021年6期)2021-11-22
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
防爆电机(2020年5期)2020-12-14
中学生数理化·八年级物理人教版(2019年6期)2019-06-25
电测与仪表(2016年14期)2016-04-11
