
酸水解条件对大黄及其制剂中蒽醌类成分含量的影响研究
2010-09-17 01:32封淑华韩桂茹
中成药 2010年7期
封淑华, 韩桂茹, 冯 丽
(河北省药品检验所,河北石家庄050011)
大黄是一种临床上应用最广泛的中药,主含蒽醌类成分,以蒽醌苷及其苷元存在于植物体内。药理研究表明:游离大黄蒽醌具抗菌、消炎、止血等作用;结合大黄蒽醌具较强的泻下作用[1]。在中药成方制剂中有采用大黄泻下功效的[2],有采用其抗菌、消炎、止血功效的。其质量控制方法目前为止都是测定的水解后的大黄总蒽醌,该指标无法区分制剂的泻下和抗菌、消炎功效。笔者试采用同一流动相,分别测定制剂中的总蒽醌、游离蒽醌和结合蒽醌的含量,按功效分别控制制剂的结合与游离蒽醌含量,实现了测定指标与功效结合的愿望。但在测定过程中发现同一样品,游离大黄酸的含量大于总大黄酸含量。对此,进行探讨研究,判明了发生的原因。报道如下:
1 仪器与试药
Waters2695高效液相色谱仪(美国 Waters公司)。
大黄素(批号:110756—200110)、大黄酚(批号:110796—200513)、大黄酸 (批号:110757—200206)、大黄素甲醚(批号:110758—200509)、芦荟大黄素(批号:110795—200504),购自中国药品生物制品检定所;三黄片(批号:080202)由河南宛西制药厂提供;麻仁润肠丸(批号:7013195)由北京同仁堂药厂提供。乙腈、甲醇、磷酸为色谱纯,其它试剂均为分析纯。
2 方法与结果
2.1 色谱条件 Shimadzu C18色谱柱(4.0 mm×250 mm,4.5 μm),流动相为乙腈-甲醇-0.1%磷酸(42∶23∶35),流速1.0 mL/min。检测波长254 nm。
2.2 对照品溶液的制备 精密称取大黄素、大黄酚、大黄酸、大黄素甲醚、芦荟大黄素适量,分别加甲醇溶解制成浓度为每1 mL含5~10μg的溶液,摇匀,即得。
2.3 供试品溶液的制备
2.3.1 总蒽醌溶液的制备 精密称取大黄药材粉末0.3 g(或三黄片粉末0.5 g、麻仁润肠丸小碎粒1 g),分别精密加入不同比例的甲醇-盐酸溶液25 mL,称重,麻仁润肠丸浸泡10 h以上,超声使溶散,置80℃水浴中回流30 min,放冷至室温,补足重量,摇匀,滤过,精密量取续滤液1 mL(麻仁润肠丸2 mL),至10 mL(麻仁润肠丸5 mL)量瓶中,加2%的氢氧化钠溶液0.5~1 mL,加甲醇定容至刻度,摇匀,滤过,取续滤液作为测定总蒽醌的供试品溶液。
2.3.2 游离蒽醌溶液的制备 精密称取大黄药材粉末0.1 g(或三黄片粉末0.3 g、麻仁润肠丸小碎粒0.8 g),精密加甲醇25 mL,麻仁润肠丸浸泡10 h以上,用玻棒研磨使样品溶散,用数滴甲醇冲洗玻棒于锥形瓶中,超声处理(功率160 W,频率50 kHz)30 min,放冷,再称定重量,用甲醇补足或挥散至原重量,摇匀,滤过,取续滤液作为测定游离蒽醌的供试品溶液。
2.4 测定 精密量取对照品溶液和供试品溶液各5~10μL,注入液相色谱仪,记录色谱图,色谱峰面积。对照品溶液和大黄药材溶液总蒽醌色谱图、游离蒽醌色谱图见图1~图3;不同酸浓度下的三黄片总蒽醌和游离蒽醌各峰面积见表1、各成分的变化趋势,见图4、5、6;不同酸浓度下的麻仁润肠丸总蒽醌和游离蒽醌各峰面积见表2、各成分的变化趋势,见图 7、8。

图1 大黄5种对照品HPLC色谱图

图2 大黄药材中总大黄蒽醌HPLC色谱图

图3 大黄药材中游离大黄蒽醌HPLC色谱图
从图1~图3,清楚看到大黄中5种蒽醌成分分离良好,总大黄蒽醌样品中的波峰数多于游离大黄蒽醌样品中波峰数,而且总大黄蒽醌中的波峰2与波峰6,在不同酸度下发生相互转变,即波峰2随着酸浓度的减小,峰面积升高,波峰6随着酸浓度的减小,峰面积在缩小,且不同酸浓度下的波峰2和波峰6面积相加,近于一固定值。
3 讨论
3.1 从图2、3得知,总大黄蒽醌中的波峰2(大黄酸)与波峰6,在不同酸度下发生相互转变,二峰面积之和基本是一定值,5个已知组分可以准确计算含量,而波峰6成分未知,无法计算含量,在控制称样量几乎一致的情况下,选择以峰面积表示其相对变化趋势更为直观。

表1 不同酸解条件下三黄片中各蒽醌成分的峰面积值

图4 不同酸浓度下三黄片中芦荟大黄素、大黄素、大黄酚、大黄素甲醚的峰面积变化趋势图

图5 不同酸浓度下三黄片中大黄酸、波峰6的峰面积变化趋势图
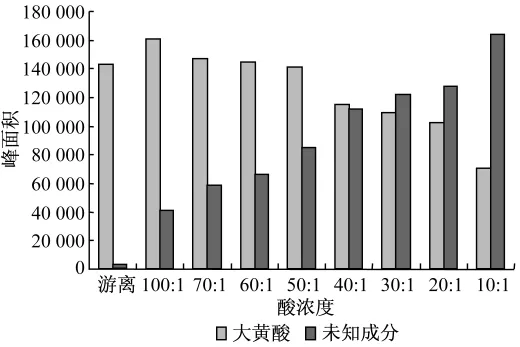
图6 不同酸浓度下三黄片中大黄酸、波峰6的峰面积变化趋势柱形图

图7 不同酸浓度下麻仁润肠丸中芦荟大黄素、大黄素、大黄酚、大黄素甲醚的峰面积变化趋势图
3.2 通过上述探讨研究得知,大黄或含大黄的制剂在同时测定总大黄蒽醌与游离大黄蒽醌时,水解酸度对芦荟大黄素、大黄素、大黄酚、大黄素甲醚的波峰面积几乎无影响,也就是酸度大小不影响上述四成分的含量,但对大黄酸的影响却比较严重。从表1的数值看到要使三黄片中总大黄酸大于游离大黄酸,甲醇-盐酸的浓度要在60∶1~100∶1之间;从表2的数值看到,麻仁润肠丸中,即便甲醇∶盐酸的浓度减小到100∶1,总大黄酸的浓度仍然比游离的低,是一负值。提示由于组方不同,影响因素而不一样,如要以大黄酸为指标进行含量测定,必须具体品种具体考察酸水解浓度。
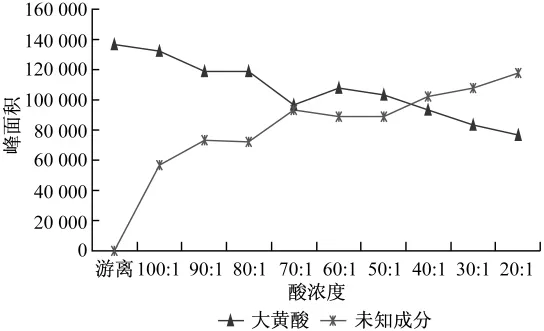
图8 不同酸浓度下麻仁润肠丸中大黄酸、波峰6的峰面积变化趋势柱形图
3.3 从表1、2的测定数值还了解到,在不同酸度下三黄片和麻仁润肠丸中的大黄酸都是随着酸浓度的减小,峰面积增大,同时与其相对应的未知峰6,则随着酸浓度的减小而减小,二峰面积之和基本是一定值,三黄片中二峰之和在20~23万之间变化(因取样量稍有差异);麻仁润肠丸中二峰之和在18~19万之间变化。数据提示:大黄酸随着酸度升高,进一步水解为极性小的成分。为确定其是否同一母核化合物,对大黄酸和波峰6,采用二极管阵列检测器,进行了光谱扫描,结果证明二峰为同一母核化合物,大黄酸进一步分解为波峰6。光谱扫描见图9。

表2 不同酸解条件下麻仁润肠丸中各蒽醌成分的峰面积值

图9 大黄酸与未知成分的光谱扫描图
3.4 通过对中药代表性片剂和蜜丸的酸水解浓度考察,发现三黄片随着酸浓度的减小,水解后的样品过滤速度减慢,到甲醇-盐酸为100∶1时,几乎无法过滤;但麻仁润肠丸则无此情况产生,分析是片剂中的辅料影响了过滤速度。
3.5 从被测定成分的保留和运行时间、波峰面积大小、分离情况以及影响因素等综合考虑,认为在中药成方制剂中如若以大黄为含量测定目标药材,以大黄素和大黄酚为测定指标比较适宜。二者波峰面积占5种成分面积之和的约60% ~70%,而且不受酸水解浓度影响,运行时间可调整在20~30 min之内,大黄素、大黄酚和大黄素甲醚波峰分离良好。
3.6 目前各类国家标准制剂中的大黄均是以大黄素、大黄酚为指标进行含量测定的[2],上述研究证实了其测定指标的合理性。
[1]乔传卓,张汉明,宓鹤鸣.生药学[M].上海:同济大学出版社,1995:184-186.
[2]中国药典[S].一部,2005:604-605.
猜你喜欢
检察风云(2022年10期)2022-06-02
水利规划与设计(2020年1期)2020-05-25
健康大视野(2019年16期)2019-08-09
中成药(2018年10期)2018-10-26
天然产物研究与开发(2018年8期)2018-09-10
中成药(2018年4期)2018-04-26
中国粮油学报(2017年12期)2018-01-10
中国医药指南(2016年1期)2016-07-11
