
配合物[Cu(m-TFBA)(phen)(H2O)2]·(m-TFBA)的水热合成、晶体结构及量子化学研究
2010-11-10 01:00季宁宁石智强赵仁高
无机化学学报 2010年6期
季宁宁 石智强 赵仁高
(1泰山学院化学与环境科学系,泰安 271021)(2泰山学院材料与化学工程系,泰安 271021)
配合物[Cu(m-TFBA)(phen)(H2O)2]·(m-TFBA)的水热合成、晶体结构及量子化学研究
季宁宁1石智强*,2赵仁高2
(1泰山学院化学与环境科学系,泰安 271021)(2泰山学院材料与化学工程系,泰安 271021)
以醋酸铜、间三氟甲基苯甲酸(m-TFBA)和邻菲咯啉(phen)为原料在甲醇水介质中通过水热反应,合成了一个新的单核铜配合物[Cu(m-TFBA)(phen)(H2O)2]·(m-TFBA),用元素分析、红外光谱和热重分析对配合物进行了表征。X-射线单晶衍射表明,配合物属三斜晶系,空间群 P1,晶胞参数:a=1.00161(10)nm,b=1.15069(12)nm,c=1.28649(12)nm,α=82.217(2)°,β=84.767(2)°,γ=66.371(2)°,V=1.3448(2)nm3,Z=2,Dc=1.625 g·cm-3,R1[I>2σ(I)]=0.0421,wR2[I>2σ(I)]=0.0958。 铜分别与来自邻菲咯啉的 2 个氮原子、间三氟甲基苯甲酸的1个氧原子和2个水分子中的2个氧原子配位,形成变形的四方锥结构。配合物通过强的O-H…O氢键作用形成了二聚体结构,该二聚体又通过分子间弱的C-H…O氢键作用形成了一维链状结构。配合物中配位的间三氟甲基苯甲酸上的三氟甲基基团具有无序结构。对配合物中[Cu(m-TFBA)(phen)(H2O)2]+进行了量子化学从头计算,探讨了配合物的稳定性、分子轨道能量以及一些前沿分子轨道的组成特征。
间三氟甲基苯甲酸;邻菲咯啉;铜配合物;晶体结构;从头计算
0 引 言
铜是生物体中必须的微量元素,在生命过程中起着传递电子、输送氧、以及清除超氧负离子等重要作用[1]。由羧酸与金属离子构筑的配合物多数具有丰富的结构类型和特殊的性能,在材料、药物、分子电化学、生物化学、生物制药等许多领域中表现出了潜在的应用价值[2-10]。芳香羧酸类配体苯甲酸及其衍生物在结构上具有一定的刚性和稳定性,在芳环上引入不同的取代基会对配合物的结构和性能会产生不同程度的影响[11],对于以位阻较大的间三氟甲基苯甲酸做配体的配合物报道的较少,因此为了进一步探索芳香羧酸类配体与相应配合物结构和性能之间的关系,我们以间三氟甲基苯甲酸为配体,邻菲咯啉为第二配体,通过水热反应合成了与Cu的三元配合物,通过元素分析、红外光谱和热重分析对其进行了表征,并利用X-射线单晶衍射测定了其晶体结构。并对其结构进行了量子化学从头计算,探讨了配合物的稳定性、分子轨道能量以及一些前沿分子轨道的组成特征。
1 实验部分
1.1 仪器与试剂
元素分析用PE-2400Ⅱ型元素分析仪测定;红外光谱用Nicolet 6700型红外光谱仪 (KBr压片)测定;热重分析用METTLER TGA/DSC 1100型热重分析仪测定;晶体结构用Bruker Smart APEXⅡCCD型单晶测试仪测定。
所用试剂均为分析纯试剂。
1.2 配合物的合成
先将 1.0 mmol Cu(Ac)2·H2O、2.0 mmol间三氟甲基苯甲酸和1.0 mmol邻菲咯啉溶于15 mL甲醇水(V甲醇∶V水=2∶1)溶液中,并用稀 NaOH 调节溶液至pH=5.0~6.0,室温下搅拌此混合溶液 0.5 h 后,将反应混合物转移到25 mL不锈钢反应釜中,于140℃反应72 h。自然降温至室温后,开釜后得到绿色晶体。产率 38%(以元素 Cu计)。元素分析按C28H20CuF6N2O6,计算值(%):C 51.11,H 3.06,N 4.26;实测值(%):C 50.95,H 3.18,N 4.34。 IR 主要吸收峰(cm-1)为:3406,1644,1547,1426,1396,1020,850,787,666,492。
1.3 晶体结构测定
选取尺寸为 0.15 mm×0.12 mm×0.10 mm 的晶体置于Bruker Smart APEXⅡCCD型X射线单晶衍射仪上,以石墨单色化的 Mo Kα(λ=0.071073 nm)辐射为光源,在室温(295±2)K下,以φ~ω扫描方式扫描, 在 1.60°≤θ≤25.04°范围内共收集 7 164 个强衍射点,其中独立衍射点4725个(Rint=0.0222),I>2σ(I)的可观测衍射点为3736个。晶体结构由直接法解出,非氢原子的坐标是在以后的数轮差值Fourier合成中陆续确定的。基于F2用SHELXL-97[12]程序对全部非氢原子的坐标及各向异性参数进行最小二乘法精修。碳上的氢原子根据理论加氢获得。 最终偏离因子 R1=0.042 1,wR2=0.095 8;w=1/[σ2(Fo2)+(0.044 4P)2+0.719 7P],其中 P=(Fo2+2Fc2)/3;(Δρ)max=390 e·nm-3,(Δρ)min=-507 e·nm-3。 配合物的晶体学数据列于表1。
CCDC:735634。

表1 配合物的晶体结构数据Table 1 Crystal data and structure refinements of the title complex

续表1
2 结果与讨论
2.1 配合物的红外光谱
以KBr压片,在4000~400 cm-1范围内测定了配合物的IR谱。配合物在3406 cm-1附近有一强而宽的吸收峰,它是配合物中水分子的特征吸收峰;配合物中羰基的不对称伸缩νas(COO-)及对称伸缩νs(COO-)振动的特征吸收峰分别出现在1 644和1396 cm-1处,其 Δ[νas(COO-)-νs(COO-)]=248 cm-1,大于200 cm-1,表明配合物中间三氟甲基苯甲酸根的羧基氧以单齿形式与Cu配位[13],同时在1 020 cm-1出现了C-F特征吸收峰。配合物中邻菲咯啉的特征吸收峰也发生了移动:从1 421、853和739 cm-1处的吸收峰分别移动到了 1 426、850和787 cm-1处,说明邻菲咯啉的氮原子与Cu发生了配位。红外分析结果与晶体结构分析结果一致。
2.2 晶体结构
配合物的分子结构见图1,二聚体结构见图2,一维链状结构见图3,主要键长和键角列于表2,主要氢键列于表3。

图1 标题配合物的分子结构Fig.1 Molecular structure of the title complex

图2 标题配合物的二聚体结构图Fig.2 Dimer structure formed by hydrogen bonds of the title complex

图3 配合物的一维链结构Fig.3 1D chain structure of the title complex
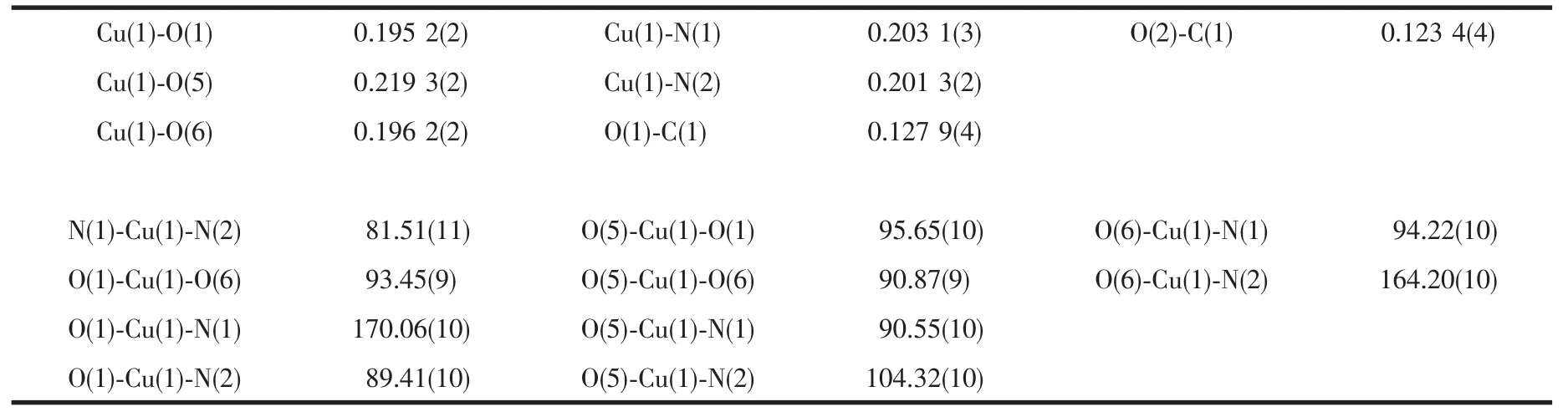
表2 配合物的主要键长和键角Table 2 Selected bond lengths(nm)and angles(°)for the title complex

表3 配合物的氢键键长和键角Table 3 Hydrogen bond lengths and angles for the title complex
由图2可知,配合物分子通过分子内氢键O-H…O 氢键:O(5)-H(1)…O(3),O(6)-H(4)…O(2)和分子间 O-H…O 氢键:O(5)-H(2)…O(3A),O(6)-H(5)…O(4A)作用形成了二聚体结构。由图3可以看出二聚体结构又通过分子间弱的C(7)-H(7)…O(2B)氢键作用形成了一维链状结构。
2.3 热重分析
在空气气氛中,升温速率为5℃·min-1,测定了标题配合物的TG曲线,在室温至700℃范围内配合物的失重分为2个阶段进行,在70~120℃之间配合物失重5.86%,对应于失去配合物中2个配位的水分子(理论值为 5.48%);从 170~400 ℃之间配合物失重81.94%,对应于失去配合物中的2个间三氟甲基苯甲酸和邻菲咯啉分子(理论值为82.43%),在这一过程中,配合物分子骨架崩塌。由于是在空气中,所以标题配合物经过复杂的分解氧化最终转化为稳定的氧化铜,最后的残余物残留率为12.20%(理论值为 12.09%)。
2.4 配合物的能量和分子轨道组成
根据配合物晶体结构的各原子坐标位置,取其中的[Cu(m-TFBA)(phen)(H2O)2]+结构单元,在B3LYP/lanl2dz基组水平上,进行了量子化学单点计算。计算涉及46个原子,315个原子基函,842个初始高斯函数,其中有114个占据轨道。所有计算均运用Gaussian 98W[19]程序包完成。
计算得到体系总能量为-1 677.650 9 a.u.,占据轨道的能量HOMO为-0.365 0 a.u.,LUMO的能量为-0.2137 a.u.。由于体系能量与占据前沿轨道的能量均较低,表明配合物基态较为稳定,这与实验结果是一致的。从氧化还原或电荷转移的角度分析,由于HOMO能级较低,并且两前沿轨道间的能量间隙达到了0.15079 a.u.,说明该配合物难以给出电子被氧化。
为探索该配合物的电子结构与成键特征,对配合物分子轨道进行了系统分析[20],用参与组合的各类原子轨道系数的平方和表示其在分子轨道中的贡献,并经归一化。把配合物原子分为七部分:(a)Cu原子;(b)N原子;(c)羧基氧O原子;(d)水上的氧O原子;(e)F原子;(f)邻菲咯啉环C原子;(g)间三氟甲基苯甲酸环C原子;(h)氢原子H。分别讨论了5个前沿占据轨道和5个前沿非占轨道,计算结果如表4和图4所示。
由表4和图4可知,配合物各原子轨道对分子轨道的贡献,在较深的占有分子轨道变化比较复杂,较深的非占分子轨道变化较小,在前沿轨道及其附近有较明显变化,限于篇幅,仅重点讨论前沿分子轨道的情况。

表4 配合物的分子轨道组成Table 4 Some calculated frontier molecular orbital composition(%)of the title complex

图4 配合物的前沿分子轨道示意图Fig.4 Schematic diagram of the frontier MO for the title complex
2.5 电子结构布居分析
由Mulliken布局分析得到的结构单元的原子净电荷如表5所示。结果表明:铜原子、氢原子以及不连氢的碳原子均带正电荷,中心铜原子失去较多电子而荷0.692 057的正电;与氢相连的碳原子、氮原子、氧原子和氟原子均带负电荷。特别应该指出的是负电荷主要集中在与铜原子直接键连的周围原子上,分别达到 N(1):-0.263 929,N(2):-0.273 385,O(1):-0.495 355,O(5):-0.706 632,O(6):-0.767022,这表明电荷通过Cu-N和Cu-O键传递给了C原子与O原子,因此这些键极性较强。

表5 配合物的原子电荷分析(B3LYP/Lanl2dz)Table 5 Atomic charge populations at B3LYP/Lanl2dz level
[1]SUN Wei-Yin(孙为银).Coordination Chemistry(配位化学).Beijing:Chemical Industry Press,2004.
[2]XU Gui-Ji(徐贵基),PAN Zhao-Rui(潘兆瑞),ZHENG He-Geng(郑和根),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25(9):1551-1556
[3]Kim J C,Lough A J,Hyejeong J.Inorg.Chem,Commun.,2002,5(8):616-620
[4]YANG Ying-Qun(杨颖群),LI Chang-Hong(李昶红),LI Wei(李 薇),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2008,24(4):510-514
[5]LI Rong(历 荣),CHEN Peng-Gang(陈鹏刚).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2008,24(4):657-660
[6]LI Dong-Ping(李东平),CHEN Zhi-Min(陈志敏),KUANG Yun-Fei(匡云飞),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2007,23(5):892-896
[7]LI Wei(李 薇),LI Chang-Hong(李昶红),YANG Ying-Qun(杨颖群),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2007,23(7):1264-1268
[8]Ono K,Yoshizawa M,Akita M,et al.J.Am.Chem.Soc.,2009,131:2782-2783
[9]ZHANG Jing(张 静),ZHANG Li-Ping(张丽萍),ZHU Long-Guan(朱龙观).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2008,24(1):27-31
[10]LI Ye(李 野),WANG Ru(王 茹),NIU Shu-Yun(牛淑云),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2008,24(11):1753-1760
[11]CHEN Zhi-Min(陈志敏),ZHANG Fu-Xing(张复兴),ZENG Rong-Ying(曾荣英),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25(3):548-551
[12]Sheldrick G M.SHELXL-97,Program for the Solution and the Refinement of Crystal Structures,University of Gottingen,Germany,1997.
[13]Nakamota K,Translated by HUANG De-Ru(黄德如),WANG Ren-Qing(汪仁庆).Infrand and Raman Spectra of Inorganic and Coordination Compounds.3rd Ed.(无机和配位化合物的红外和拉曼光谱.3版).Beijing:Chemical Industry Press,1986.
[14]Chui S S Y,Shek L Y.J.Am.Chem.Soc.,2000,122(26):6293-6294
[15]TIAN Chun-Liang(田春良).Chemical Reagents.(Huaxue Shiji),2008,30(8):591-593
[16]Ranford J D,Sadler P J,Tocher D A.J.Chem.Soc.,Dalton Trans.,1993:3393-3399
[17]Kaizer J,Csay T,Speier G,et al.Inorg.Chem,Commun.,2006,9(10):1037-1039
[18]LI Chang-Hong(李昶红),HE Xiao-Mei(何晓梅),YANG Ying-Qun(杨颖群),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2007,23(8):1449-1452
[19]Aeleen F,Michael J F.Gaussian 98 User′s Reference,Gaussian,Inc.,Garnegie Office Park,Bldg.6 Pittsburgh,PA15106 USA
[20]KUANG Dai-Zhi(邝代治),FENG Yong-Lan(冯泳兰),WANG Jian-Qiu(王剑秋),et al.Computers and Applied Chemistry(Jisuanji Yu Yingyong Huaxue),2006,23(12):1188-1192
Hydrothermal Synthesis,Crystal Structure and Quantum Chemistry of the Complex[Cu(m-TFBA)(phen)(H2O)2]·(m-TFBA)
JI Ning-Ning1SHI Zhi-Qiang*,2ZHAO Ren-Gao2
(1Department of Chemistry and Environment Science,Taishan University,Taian,Shandong 271021)(2Department of Materials science and Chemical Engineering,Taishan University,Taian,Shandong 271021)
The tiltle complex[Cu(m-TFBA)(phen)(H2O)2]·(m-TFBA)(m-TFBA=m-trifluoromethylbenzoic acid,phen=1,10-phenanthroline)was hydrothermally synthesized by the reaction of cupric acetate monohydrate with m-TFBA and phen in the solvent mixture of methanol and water.The title complex was characterized by elemental analysis,IR spectra and TG.Its crystal structure was determined by X-ray single crystal diffraction study.The crystal belongs to triclinic with space group P1,a=1.00161(10)nm,b=1.15069(12)nm,c=1.286 49(12)nm,α=82.217(2)°,β=84.767(2)°,γ=66.371(2)°,V=1.3448(2)nm3,Z=2,Dc=1.625 g·cm-3,R1[I>2σ(I)]=0.0421 and wR2[I>2σ(I)]=0.0958.The copperion is five-coordinated with two nitrogen atoms from one phen,one oxygen atom from one m-TFBA and two water oxygen atoms,forming a distorted square-pyramidal configuration.The-CF3group from the ligand m-TFBA shows disorder in the title complex.A dimer structure was formed by strong O-H…O hydrogen bonds.Weak intermolecular C-H…O hydrogen bonds link the molecules into one-dimensional chain.The study on the[Cu(m-TFBA)(phen)(H2O)2]+cation has been performed with quantum chemistry calculation bymeans of G98W package and taking Lanl2dz basis set.The stabilities of the complex,the orbital energies and composition characteristies of some frontier molecular orbitals have been investigated.CCDC:735634.
m-trifluoromethylbenzoic acid;1,10-phenanthroline;Coppercomplex;crystal structure;ab initio method
O614.121
A
1001-4861(2010)06-1025-06
2009-12-30。收修改稿日期:2010-03-25。
泰山学院人才引进项目(No.Y06-2-08)资助。
*通讯联系人。 E-mail:kobeecho@163.com
季宁宁,女,30岁,讲师;研究方向:功能配合物的合成与分析。
猜你喜欢
云南化工(2020年11期)2021-01-14
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
百科知识(2016年22期)2016-12-24
衡阳师范学院学报(2016年3期)2016-07-10
当代化工研究(2016年5期)2016-03-20
中学化学(2015年12期)2016-01-19
火炸药学报(2014年3期)2014-03-20
原子与分子物理学报(2014年3期)2014-02-28
无机化学学报(2014年8期)2014-02-28
无机化学学报(2014年1期)2014-02-28
