
硝呋太尔合成工艺的优化
2011-12-27 01:05尚红艳陈亚鹏
河北省科学院学报 2011年2期
尚红艳,陈亚鹏
(1.石家庄市中小学生校外综合实践活动基地,河北,石家庄 050000;2.河北化工医药职业技术学院,河北,石家庄 050026)
硝呋太尔合成工艺的优化
尚红艳1,陈亚鹏2
(1.石家庄市中小学生校外综合实践活动基地,河北,石家庄 050000;2.河北化工医药职业技术学院,河北,石家庄 050026)
对已有硝呋太尔合成工艺进行改进,直接采用市售的甲硫醇钠为原料,与环氧氯丙烷缩合得到甲硫基环氧丙烷。甲硫基环氧丙烷经过肼解、环合、缩合得到硝呋太尔。其中,肼解反应得到的肼解物不经分离直接进行环合反应,硝呋太尔收率比国内外文献报道高10%左右,降低了生产成本,适合工业化生产。
硝呋太尔;合成工艺;优化
硝呋太尔[1](Nifuratel),化学名为5-[(甲硫基)甲基]-3-[(5-硝基亚糠基)氨基]-2-噁唑烷酮是由意大利普利(PoLi)化学工业公司独家开发生产的一个主要用于妇科混合性感染的新药。
硝呋太尔的合成有以下两种方法:一是甲硫醇与环氧氯丙烷缩合得到甲硫基环氧丙烷,甲硫基环氧丙烷经肼解、环合和缩合得到硝呋太尔[2]。本方法缺点是副反应多,且甲硫醇不易运输,收率也不高。二是硫酸二甲酯与硫脲反应得到甲基异硫脲,加碱水解后与环氧氯丙烷反应得到甲硫基环氧丙烷,甲硫基环氧丙烷经肼解、环合和缩合得到硝呋太尔[3]。本方法优点是副反应减少,收率有所提高,避免了甲硫醇运输的问题,缺点是要用到剧毒物硫酸二甲酯。
针对以上问题,笔者对合成工艺进行了改进,采用市售的甲硫醇钠为原料,与环氧氯丙烷缩合得到甲硫基环氧丙烷,甲硫基环氧丙烷经过肼解、环合、缩合得到硝呋太尔,其中,肼解反应得到的肼解物不经分离直接进行环合反应,反应收率比国内外文献报道高10%左右[4,5,6]。
1 实验部分
硝呋太尔合成路线:
(1)环氧丙甲硫醚的制备

(4)水解
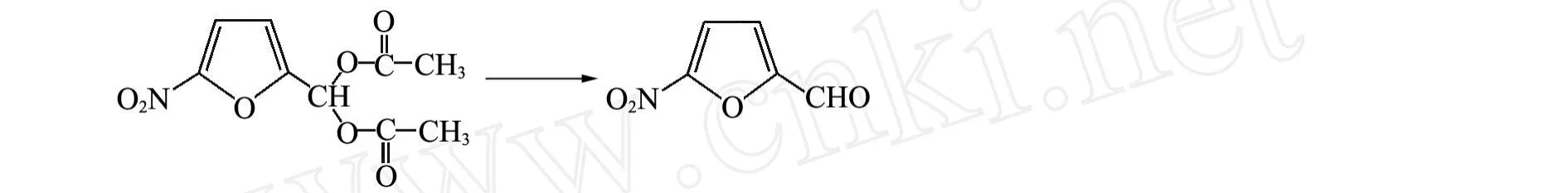
(5)缩合

1.1 仪器与试剂
电动搅拌器:2003型;加热套:型号98-I-B型;恒温干燥箱:型号202型。
甲硫醇钠(20%);环氧氯丙烷;二氯甲烷;四丁基溴化铵;水合肼(80%)碳酸二乙酯;甲醇钠;无水甲醇;无水乙醇;5-硝基-2-糠醛二醋酸酯;95%乙醇;10%硫酸(硫酸35.2g纯化水316.8g)。
1.2 环氧丙甲硫醚的制备
将环氧氯丙烷84m l、二氯甲烷280m l、四丁基溴化铵12g按工艺量投入1000m l反应瓶中。开启搅拌,在25℃以下滴加甲硫醇钠425g,并控制温度不超过25℃。滴加时间2h左右(滴加要缓慢、均匀)。滴加完毕,在15-25℃保温2h,反应液始终呈白色混浊状态。反应结束后,静置20min左右,分层,分出下层二氯甲烷相,上层水相40m l二氯甲烷萃取两次,静置分层,称量水相重量,合并二氯甲烷相,用纯化水洗至中性(洗2-3次,每次100m l)每次洗涤完毕用试纸测水相p H值,最后水洗液p H值应达到中性。静置分层20min后分出有机相,有机相加入15g M gSO4干燥2-4h(该时间不完全确定)。减压蒸馏除去二氯甲烷,收集bp20 50~55℃馏分,得78.5g产物,收率为66.8%。n25D=1.4815。
1.3 肼解
先将水合肼125m l投入到1000m l三口瓶中,搅拌升温到95℃左右。缓慢滴加环氧丙甲硫醚68.39g,滴加温度控制在100℃以下(温度过高会导致部分水合肼挥发),滴加时间1h左右。在100℃下(95-98℃)保温3h(为避免水合肼蒸发,在反应体系上加回流冷凝)。反应完毕,降至室温(25-30℃),将常压装置改为减压装置,观察瓶内料液沸腾和出馏情况,没有液体冷凝时停止加热,剩余产物直接环合,产物约90g。
1.4 环合
先将碳酸二乙酯77.5g加入到肼解物89.4g中搅拌,呈白色混浊。将甲醇钠10g溶于甲醇加入到反应体系中(要确保甲醇钠有效的溶解到甲醇中)。加热回流2h,反应液颜色为浅咖啡色。瓶内温度73 -76℃左右。在反应过程中观察反应液是否在瓶壁上产生一定量的白色固体,如果有,证明甲醇钠分解,从而影响环合反应。加热回流15m in左右,取少量以前保留的水解物于试管中,取少量环合物滴加到水解物中环合物和水解物按1:3的比例加入,观察如果有大量的黄色沉淀证明环合反应充分,可进行下一步反应,如料液呈深红色而且黄色沉淀析出很少或者较长时间才有析出,证明环合反应不充分。反应完毕,常压蒸出甲醇、乙醇,此处的目的是使体系反应更加充分。在0.03Mpa真空度条件下进一步蒸除甲醇、乙醇。体系冷却到35℃左右,加入100m l无水乙醇稀释,料液呈浅咖啡色(有隐约兰绿色)。
1.5 水解
把5-硝基-2-糠醛二醋酸酯176g,95%乙醇441m l,10%硫酸352m l(硫酸35.2g纯化水316.8g)投入到反应瓶中,加热回流至物料溶解。64-68℃物料溶解,80℃大量回流。回流30m in。置于3-5℃下静置分层,12h左右,取上层黄色液体待用。
1.6 缩合
在室温下(25-30℃)下,将环合物106.5g缓慢滴加到水解物92.7g中(要求避光滴加),开始体系迅速变成深红色,而后开始有大量的黄色物析出。在30-40℃下保温搅拌4h。搅拌条件下,缓慢降温至室温(20℃左右),用冷却水降温达5℃左右。抽滤(滤液保留)。滤饼用温水泡洗两次,每次700m l,利用机械搅拌均匀洗涤。洗涤目的是:洗去无机盐杂质及残留滤液。用95%乙醇550m l泡洗一次,抽滤。在79℃-81℃条件下干燥12h左右,得粗品90g左右,收率48%左右。
1.7 精制
首先要将硝呋太尔粗品研细。将冰醋酸600m l和硝呋太尔100g依次加入三口瓶中。搅拌升温至回流,110℃左右有大量回流,回流0.5h。自然降温20℃左右,用冷却水降温至10-15℃左右,过滤,尽量抽干。加入95%乙醇500m l,搅拌洗涤,过滤,尽量抽干。79-81℃避光干燥,12h左右。得精品87. 5g。精制收率85%-90%左右。高效液相色谱外标法计算,测定结果为99.6%,符合国家规定。
2 讨论
(1)多次实验结果显示,在第一步甲硫基环氧丙烷的制备中,用20%的甲硫醇钠的水溶液与环氧氯丙烷反应,采用二氯甲烷作溶剂,以四丁基溴化铵做相转移催化剂,以碘化钾为环氧氯丙烷中氯原子活化剂,得到的甲硫基环氧丙烷收率较高,可达75%以上,比文献报道要高10%以上。
(2)在第二步肼解反应中,按以往的文献的方法,所得的肼解物在减压蒸除水合肼后,需要进一步在高真空下蒸馏提纯得到肼解物(一般真空度在1mm Hg时沸点在135℃)。而按此方法在实际操作中却有很大的困难,一方面1mm Hg左右的高真空较难达到,尤其在放大实验中;另一方面,高温下所得的肼解产物颜色加深,收率下降,对最终产物硝呋太尔的外观和质量都有一定的影响,分析可能是肼解物高温下分解。经过多次实验摸索,将第二步反应所得反应液减压蒸除水合肼,所得油状物直接用于下一步反应,不仅可避免高真空操作,也可避免产物的高温分解。在蒸馏过程中,物料必须在搅拌条件下进行蒸馏,才能有效保证水合肼的彻底蒸除。从实验结果上看,收率有较大幅度的提高。
(3)合成中使用的水合肼、乙醇和冰醋酸都可通过蒸馏或精馏回收利用,回收率75%左右。产生的废水经中和后排污。固体废渣如活性炭等集中统一处理。本工艺条件下无废气产生。
3 结论
(1)整个合成过程副反应少,总收率比文献报导的高10%以上。
(2)反应条件不苛刻,不仅避免了甲硫醇运输和使用过程的问题,也避免了使用剧毒物硫酸二甲酯,使操作更方便、更安全。
(3)肼解物不经分离直接进行下一步反应,减少了肼解物在分离过程对高真空的要求,也避免了肼解物在高温蒸馏过程中的部分变质。
(4)环氧氯丙烷经取代、肼解、环合和缩合四步反应,制得硝呋太尔,其中,肼解物、环合物不经分离直接进行下一步反应,减少了分离操作步骤。
从以上几点可以看出,本工艺操作更容易、更安全,而且收率更高,所以更适合于工业化生产。
[1] Susan Budavari,Maryadele J.ONeil,Ann Smith.Merck Index[M].Eleventh Edition,1989,6442.
[2] R.Davies.Substituted Oxazolidinones[P].Belg,1963,635608.
[3] 李志万,尹立新,马继革.解放军药学学报[J].21,(4):296-287.
[4] Thomas K.Todsen,C.B.Pollard,Edward G.Rietz,Someβ-Hydrox-ypropyl Sulfides and their Derivatives[J].JAm Chem Soc, 1950,72:4000.
[5] Harry R.Snyder,Jr.,Frank F.Ebetino,1-Substituded 3-[(5-Nitrofurylidene)amino]-2-Imidazolidinones,J Med chem.1970, Vol.13,No4 756-758.
[6] 化工部科技情报研究所.化工产品手册有机化工原料(下册)[M].北京:化工出版社,1982.
Optim izing the syn thetic process of Nifuratel
SHANG Hong-yan1,CHEN Ya-peng2
(1.Shijiazhuang Com prehensive Practice Base for Primary&M idd le School,Shijiazhuang05000,China;2.Hebei Chem ical&Pharmaceutical Vocational Technology College,Shijiazhuang Hebei050026,China)
To imp rove the synthetic p rocess of nifuratel,it used the commercially available 3-methylthio-(1,2-epoxyp ropane)as raw materials,condensated w ith PO to obtain Ethylthio-epoxy p ropane. This compound was then treated acco rding to the know n p rocedures such as hydrazinolysis and cyclization and condensation to form nifuratel.Productsof hydrazinolysis reaction cyclizated directly w ithout separation.By imp roving the synthetic p rocess,the yield of compound nifuratel was exceeding 10%from w hat was reported.These reduced the cost of p roduction and was suitable for the nifuratel p roduction in factories.
Nifuratel;Synthetic p rocess;Op timization
TQ242.2
:A
1001-9383(2011)02-0058-04
2011-03-25
尚红艳(1975-),女,河北省灵寿县人,中学一级,主要从事生物和化学教学实践工作.
猜你喜欢
当代化工研究(2022年13期)2022-07-25
云南化工(2021年11期)2022-01-12
氯碱工业(2021年11期)2021-12-29
中国民间疗法(2021年13期)2021-08-30
化工时刊(2020年6期)2020-08-14
广州化工(2020年7期)2020-04-28
食品与生物技术学报(2018年6期)2018-09-20
中成药(2018年3期)2018-05-07
当代医药论丛(2017年3期)2017-11-30
食品研究与开发(2017年13期)2017-07-01
