
ZSM-5/MCM-41介孔硅铝分子筛担载Pd和Pt制备加氢脱硫催化剂
2012-01-29 02:10王林英王安杰胡永康
石油学报(石油加工) 2012年3期
王林英,王安杰,,李 翔,,周 峰,胡永康
(1.大连理工大学精细化工国家重点实验室,辽宁大连116012;2.辽宁省高校石油化工技术与装备重点实验室,辽宁大连116012)
油品的深度脱硫是清洁燃料生产所面临的重要课题。目前在炼油厂中,油品中有机含硫化合物的脱除仍主要采用加氢脱硫(HDS)工艺。二苯并噻吩(DBT)及其烷基取代的衍生物如4,6-二甲基二苯并噻吩(4,6-DMDBT)等是馏分油中最难脱除的含硫化合物[1]。这是由于烷基取代基的空间位阻效应,使得4,6-DMDBT等DBT的烷基取代衍生物中硫原子很难接近催化剂的活性中心。但是当这些芳香杂环含硫化合物中的一个芳环首先被加氢后,可以降低空间位阻,提高反应物分子的活性[2]。因此良好的深度加氢脱硫催化剂一般应具有较高的加氢活性。
负载型贵金属催化剂具有较高的加氢活性。研究表明,Pd/γ-Al2O3、Pt/γ-Al2O3以及Pt-Pd/γ-Al2O3等对DBT和4,6-DMDBT表现出很高的HDS活性,已经成为重要的深度加氢脱硫催化剂[3-4]。但贵金属催化剂最大的弱点是不耐硫。载体是贵金属多相催化剂的重要组成部分,一般来说,提高载体的酸性有利于提高贵金属催化剂的HDS活性和耐硫性能[5]。常见的酸性载体主要包括无定型硅铝、沸石或包含沸石的无机材料等。沸石虽然具有较高的酸性,但其微孔结构不适用于针对DBT等大分子含硫化合物的深度加氢脱硫过程。MCM-41等介孔材料尽管具有适宜的孔道尺寸和较大的比表面积,但其较弱的表面酸性和较差的水热稳定性限制了它们作为载体在贵金属加氢脱硫催化剂中的应用。笔者前期研究发现,在偏硅酸钠强碱性缓冲溶液体系中对不同种类沸石(如ZSM-5、HY、丝光沸石等)选择性地脱硅,然后在模板剂的作用下自组装,可以合成出孔壁中含有沸石结构单元的强酸性、高水热稳定性的介孔硅铝材料[6-8]。在此基础上,笔者合成了系列ZSM-5/MCM-41介孔硅铝分子筛,以其为载体制备负载型Pd和Pt催化剂,并以DBT作为模型化合物,考察它们的HDS催化活性。
1 实验部分
1.1 原料与试剂
偏硅酸钠(Na2SiO3·5H2O),工业级,天津东方红化工厂产品;十六烷基三甲基溴化氨(CTAB),工业级,南京旋光科技有限公司产品;氯化钯(PdCl2)和氯铂酸(H2PtCl4),分析纯,天津化学试剂一厂产品;十氢萘购自上海试剂分装厂,纯度大于99%。二苯并噻吩(DBT)由联苯(BP)和硫合成[9]。HZSM-5(n(SiO2)/n(Al2O3)=26,平均颗粒直径100nm)由大连理工绿源化工有限公司提供。
1.2 载体和催化剂的制备
按文献[10]中的方法制备Si-MCM-41,按文献[7]中的方法制备ZSM-5/MCM-41介孔硅铝分子筛。所得ZSM-5/MCM-41记为ZM(x),其中x代表母液中SiO2/Al2O3摩尔比。
采用浸渍法制备催化剂。将计量的载体分别加入PdCl2的盐酸溶液(pH<1)或H2PtCl6溶液中,室温下浸渍8h,120℃下烘干12h,在500℃空气中焙烧3h,得到氧化态Pd或Pt催化剂。所得催化剂分别记为Pt(或Pd)/Si-MCM-41和Pt(或Pd)/ZM(x),它们所含Pd和Pt的质量分数均为1.0%。
1.3 催化剂HDS反应活性评价
在内径为8mm的不锈钢固定床反应器中进行HDS反应。催化剂经压片、破碎至0.5~0.8mm后装填入反应器,用量0.05g。HDS反应前,用H2对氧化态催化剂进行还原,还原温度300℃,时间2h。在压力5.0MPa、质量液时空速(MHSV)54h-1、氢/油体积比850、反应温度300℃的条件下进行HDS反应。反应原料为质量分数0.8%的DBT/十氢萘溶液。采用HP-6890+型气相色谱仪测定原料和HDS反应产物的组成。色谱仪配有Agilent公司HP-5型毛细管色谱柱,固定相为5%二苯基95%二甲基聚硅氧烷。
DBT的HDS反应主要通过直接脱硫(DDS)和加氢(HYD)2条并行的反应路径实现脱硫。联苯(BP)是DDS反应路径的唯一产物,在有机含硫化合物如DBT存在的情况下,BP很难进一步加氢生成苯基环己烷(CHB)[11]。四氢二苯并噻吩(TH-DBT)和六氢二苯并噻吩(HH-DBT)是HYD反应路径脱硫的主要含硫中间体,TH-DBT和HH-DBT脱硫后生成CHB和联环己烷(BCH)。酸性载体负载的Pd和Pt催化剂既有酸中心也有金属中心,能发生加氢裂化反应(HYC),产物为苯、环己烷以及一些碳数小于6的小分子烷烃。
负载型Pd和Pt催化剂上DBT的反应产物中除脱硫产物和加氢裂化产物之外,还能检测到含硫中间体TH-DBT和HH-DBT。因此,用DBT加氢脱硫转化率(xHDS)表示催化剂的脱硫催化活性,可由式(1)计算。

式(1)中,cDBT,0和cDBT分别为反应器入口和出口处DBT的浓度,mmol/L;cTH和cHH分别为反应器出口处TH-DBT和HH-DBT的浓度,mmol/L。
催化剂DDS路径选择性可用BP的选择性(sBP)表示,(1-sBP)表示催化剂HYD路径的选择性。由于加氢裂化反应生成碳数小于6的烷烃难以准确检测,因此选用(1-sTH-sHH-sBP-sCHB-sBCH)表示催化剂加氢裂化选择性,而用加氢裂化转化率(xHYC)表示催化剂的加氢裂化催化活性,可由式(2)计算。

式(2)中,sTH、sHH、sBP、sCHB和sBCH分别表示TH-DBT、HH-DBT、BP、CHB和BCH的选择性,%。
1.4 催化剂的表征
采用Rigaku D/MAX-2400型衍射仪获取载体的XRD谱,以CuKα为辐射源,Ni滤波,管电压40kV,管电流100mA。采用ASAP 2010吸附仪测定载体的N2吸附-脱附等温线。采用EQUINOX55型红外光谱仪吡啶吸附红外光谱法测定载体酸性,吡啶脱附温度为300℃。采用SRS 3400型X射线荧光光谱仪(XRF)测定样品的Al2O3质量分数。用JEM-2100型高分辨电镜观察催化剂形貌。
2 结果与讨论
2.1 载体Si-MCM-41和ZM(x)以及其为载体的Pd和Pt催化剂的物性表征结果
2.1.1 Si-MCM-41和ZM(x)的XRD分析
图1为Si-MCM-41和ZM(x)的XRD谱。从图1看到,所有样品在2θ为2.1°附近均能够观察到很强的对应于MCM-41分子筛(100)晶面的特征衍射峰,表明合成的ZSM-5/MCM-41介孔硅铝分子筛具有良好的介孔结构;在2θ为5°~60°之间,ZM(x)没有ZSM-5分子筛的特征衍射峰,表明这些样品不是Si-MCM-41与ZSM-5的机械混合物。本课题组前期研究红外表征结果表明[7-8],合成的ZSM-5/MCM-41介孔硅铝分子筛在波数为553cm-1处能够观察到归属于分子筛晶体中高度扭曲的五元环结构(T—O—T,T=Si或Al)的吸收峰,说明ZSM-5在偏硅酸钠溶液中发生降解,主要以分子筛次级结构单元的形式存在于ZM系列的介孔孔壁中。

图1 Si-MCM-41和ZM(x)的XRD谱Fig.1 XRD patterns of Si-MCM-41and ZM(x)(1)Si-MCM-41;(2)ZM(90);(3)ZM(60);(4)ZM(50)
2.1.2 Si-MCM-41和ZM(x)的孔结构参数、Al2O3含量及B/L分析
表1列出了由N2物理吸附、XRD及XRF得到的Si-MCM41与ZM(x)的结构参数和Al2O3质量分数。从表1可以看出,ZM(x)系列样品的比表面积随其n(SiO2)/n(Al2O3)降低而降低,但介孔孔径基本不发生变化,都在2.7nm左右,低于Si-MCM-41,因为孔壁厚度比Si-MCM-41有了显著增加。通过XRF测得ZM(x)样品中n(SiO2)/n(Al2O3)低于投料的n(SiO2)/n(Al2O3),即样品组成中Al2O3含量高于反应母液,并且在n(SiO2)/n(Al2O3)较高时,富铝现象更明显。Reddy等[12]曾报道,Al-MCM-41样品中n(SiO2)/n(Al2O3)低于相应母液n(SiO2)/n(Al2O3),认为这是由于硅源和铝源自身性质的差别以及二者的比例不同导致了Al物种与MCM-41前体胶的反应活性不同,从而影响最终样品中Al的引入量。

表1 Si-MCM-41和ZM(x)的孔结构参数、Al2O3质量分数及B酸和L酸比值(B/L)Table 1 The structural parameters,Al2O3mass fraction and the molar ratios of Brønsted acid sites to Lewis acid sites(B/L)of Si-MCM-41and ZM(x)
2.1.3 Si-MCM-41和ZM(x)的FT-IR分析
图2为Si-MCM-41和ZM(x)的吡啶吸附FT-IR谱。从图2看到,Si-MCM-41在波数为1445和1596cm-1处的吸收带对应通过氢键吸附在表面硅羟基上的吡啶,这是全硅材料的特征吸附峰[13];ZM(x)在1455cm-1附近的吸收带归属为吡啶在L酸中心的吸附,1545cm-1附近的吸收带归属为吡啶在B酸中心的吸附,而波数在1490cm-1附近的强吸收带则归属为吡啶同时在B酸和L酸中心的吸附[13],在1445和1596cm-1处没有观察到归属于氢键吸附的吡啶特征峰。图2结果表明,引入ZSM-5后ZM(x)的酸性比Si-MCM-41有显著提高。ZM(x)中B酸和L酸量比值(B/L)也列于表1。由表1可以看出,B/L值随着n(SiO2)/n(Al2O3)的降低而降低。

图2 Si-MCM-41和ZM(x)的吡啶吸附FT-IR谱Fig.2 Pyridine adsorption FT-IR spectra of Si-MCM-41and ZM(x)(1)Si-MCM-41;(2)ZM(90);(3)ZM(60);(4)ZM(50)
2.1.4 Pt(或Pd)/Si-MCM-41和Pt(或Pd)/ZM(x)催化剂的TEM分析
Pt(或Pd)/Si-MCM-41和Pt(或Pd)/ZM(x)催化剂的透射电镜照片(TEM)示于图3。从图3看到,在所有的催化剂样品中,均能够清晰地观察到规整的一维介孔孔道,说明担载贵金属不会破坏载体介孔结构。Pt(或Pd)/Si-MCM-41和Pt(或Pd)/ZM(90)催化剂活性组分的颗粒尺寸分布不均匀,分散度较差。由于PdCl2不溶于水,配制浸渍液时需要先将其溶于盐酸(pH<1)得到(PdCl4)2-阴离子,H2PtCl6水溶液的pH值也在1.5左右,与SiO2等电点(1.5~2.0)接近,使得(PdCl4)2-和(PtCl6)2-等阴离子与载体表面相互作用很弱,可能是导致制备的催化剂活性组分分散度差的主要原因[14]。Pt和Pd在Al含量较高的ZM(60)和ZM(50)载体上分散良好,颗粒分布较均匀。原因可能是Al2O3等电点在pH值为7~9之间[15],呈碱性,改善了载体与浸渍液中相应阴离子的相互作用。从活性物种形貌上看,Pd在ZM(60)和ZM(50)中倾向沿孔道方向分布,而Pt则优先聚集成小颗粒状。
2.2 DBT在Pt(或Pd)/Si-MCM-41和Pt(或Pd)/ZM(x)催化剂上HDS反应活性


图4为Pd/Si-MCM-41和Pd/ZM(x)催化DBT HDS反应加氢脱硫和加氢裂化转化率以及DDS和HYD反应路径选择性随反应时间的变化。由图4可以看出,在Pd系列催化剂上DBT HDS的HYD反应路径的选择性均大于80%,说明主要通过HYD反应路径脱硫;Pd/Si-MCM-41的HDS催化活性较低,并且失活较快,反应8h时加氢脱硫转化率由初始的60%左右降至25%。这是由于Si-MCM-41酸性很弱,因此Pd/Si-MCM-41的HDS催化活性较低。Pd/ZM(x)的HDS活性和HYD选择性显著高于Pd/Si-MCM-41,其中Pd/ZM(60)具有最高的HDS活性和HYD选择性,其HDS转化率和HYD选择性分别达到93%和97%。Pd/ZM(60)还表现出良好的稳定性,在8h内没有失活。另外Pd/ZM(x)的加氢裂化催化活性与Pd/Si-MCM-41相比有明显提高,说明载体具有较强的酸性。
图5为Pt/Si-MCM-41和Pt/ZM(x)催化DBT HDS反应加氢脱硫和加氢裂化转化率以及DDS和HYD反应路径选择性随反应时间的变化。由图5可见,在Pt系列催化剂上DBT HDS反应的DDS和HYD 2条反应路径并重,Pt/ZM(x)的加氢脱硫活性、加氢反应路径选择性、加氢裂化活性及稳定性都高于Pt/Si-MCM-41催化剂;ZM(x)的最佳母液n(SiO2)/n(Al2O3)为60,在Pt/ZM(60)上DBT的加氢脱硫转化率大于80%,HYD选择性约为60%,并且催化剂在反应8h内没有观察到明显失活。
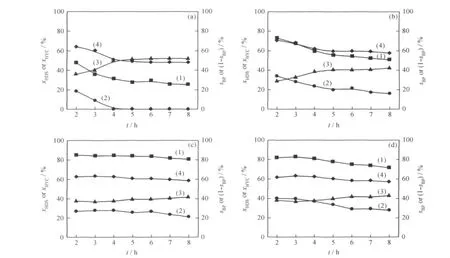
图5 Pt/Si-MCM-41和Pt/ZM(x)催化DBT HDS反应的加氢脱硫转化率(xHDS)、加氢裂化转化率(xHYC)以及DDS(sBP)和HYD选择性(1-sBP)随反应时间(t)的变化Fig.5 The HDS conversion(xHDS),the hydrocracking conversion(xHYC),the DDS pathway selectivity(sBP)and the HYD pathway selectivity(1-sBP)vs reaction time(t)in DBT HDS over Pt/Si-MCM-41and Pt/ZM(x)(a)Pt/Si-MCM-41;(b)Pt/ZM(90);(c)Pt/ZM(60);(d)Pt/ZM(50)(1)xHDS;(2)xHYC;(3)sBP;(4)1-sBP
ZM(x)和Si-MCM-41担载的Pd和Pt催化剂在DDS与HYD反应路径选择性上的差异与文献[3,16-18]报道一致,但有关机理尚存在争议。一般认为这与贵金属本身特性有关[3,16],也有认为贵金属催化剂酸性可能是影响其HDS性能重要因素。如Baldovino-Medrano等[18]通过NH3-TPD发现,Pd/γ-Al2O3酸性高于Pt/γ-Al2O3,可能是造成前者具有较高HYD选择性的重要原因之一。总的说来,ZM(x)担载的贵金属催化剂加氢活性、加氢脱硫活性及稳定性都高于Si-MCM-41担载的贵金属催化剂。根据载体和催化剂表征结果,这可能与活性组分分散度及载体酸性有关。一般认为,提高载体的酸性有利于提高催化剂HDS活性和耐硫性能[5],因为贵金属活性组分与载体表面酸中心之间存在强相互作用,活性中心部分电荷转移到载体的酸中心上,导致贵金属活性组分电子密度降低,形成具有较高加氢活性和耐硫性能的“缺电子结构”[19-21]。还有观点认为,提高载体的酸性,有利于溢流氢的生成和稳定[22-23]。氢溢流对多相催化体系有重要影响,如产生新活性中心、抑制催化剂表面积炭、提高活性组分还原能力、直接参与反应等[24]。Zhang等[25]合成了一种包覆型MCM-41/Y介-微孔分子筛,并以其作载体制备了负载型Pt-Pd催化剂,发现该催化剂在萘加氢过程中表现出较好的耐硫性能,他们认为溢流氢的“自清洁”机制对提高催化剂的性能可能起到重要作用。Sugioka等[26]则提出,杂环含硫化合物能够吸附在载体酸中心上,与贵金属表面产生的溢流氢发生反应实现脱硫,即载体酸中心本身就是催化HDS反应活性中心之一。Pt/ZM(60)和Pt/ZM(50)催化剂活性组分分散度差别不大,而Pd/ZM(50)中Pd的分散度高于Pd/ZM(60)(见图3),但Pt/ZM(60)和Pd/ZM(60)加氢脱硫反应性能均优于ZM(50)担载的催化剂(见图4和图5),这可能与B/L不同有关(见表1)。Gallezot[27]报道,B酸中心和L酸中心都可作为贵金属电子受体,诱导贵金属形成“缺电子结构”。但B酸中心与贵金属之间的相互作用可能更强,如形成金属-质子加合物(Metal-proton adduct)[28]。Guczi等[29]研究了不同预处理方法对Pt/NaY催化剂制备的影响,发现当B酸含量较高时,金属以小颗粒形式存在于Y沸石的超笼内,而当系统中仅含有L酸中心时,金属则主要以大颗粒形式分布在Y沸石表面,说明B酸中心对金属的稳定作用更强。因此,具有较高活性组分分散度和较高B/L比例的ZM(60)担载的贵金属催化剂表现出最高的加氢活性、加氢脱硫反应活性和稳定性。但需要指出的是,ZM(x)担载的贵金属催化剂也表现出较高的加氢裂化活性(见图4和图5),当用于真实油品加氢精制过程时可能会降低产品液体收率,这是酸性载体担载的贵金属加氢精制催化剂面临的重要问题之一,有待进一步研究和解决。
3 结 论
(1)通过在偏硅酸钠强碱性溶液体系中对ZSM-5降解然后在模板剂的作用下自组装,合成出具有较强酸性和不同n(SiO2)/n(Al2O3)的系列ZSM-5/MCM-41介孔硅铝分子筛ZM(x)。担载Pt和Pd后不会破坏ZM(x)介孔结构。
(2)DBT在担载的Pd催化剂上主要通过加氢路径脱硫,而在Pt催化剂上则直接脱硫和加氢2条反应路径并重。
(3)Si-MCM-41担载的Pt和Pd催化剂加氢脱硫活性和稳定性较差。当以ZM(x)作载体时,Pt和Pd催化剂加氢活性、加氢脱硫活性、加氢裂化活性及稳定性都有显著提高。ZM(x)担载的Pt和Pd催化剂加氢脱硫活性可能与其活性组分分散度,以及载体的B酸和L酸比例都有关系,具有较好的活性组分分散度和较高B/L比例的ZM(60)担载的Pd和Pt催化剂表现出最佳的加氢脱硫活性和稳定性。
[1]WHITEHURST D D,ISODA T,MOCHIDA I.Present state of the art and future challenges in the hydrodesulfurization of polyaromatic sulfur compounds[J].Adv Catal,1998,42:345-471.
[2]KABE T,ISHIHARA A,ZHANG Q.Deep desulfurization of light oil Part 2Hydrodesulfurization of dibenzothiophene,4-methyldibenzothiophene and 4,6-dimethyldibenzothiophene[J].Appl Catal A,1993,97(1):L1-L9.
[3]NIQUILLE-RÖTHLISBERGER A,PRINS R.Hydrodesulfurization of 4,6-dimethyldibenzothiophene and dibenzothiophene over alumina-supported Pt,Pd,and Pt-Pd catalysts[J].J Catal,2006,242(1):207-216.
[4]RÖTHLISBERGER A,PRINS R.Intermediates in the hydrodesulfurization of 4,6-dimethyl-dibenzothiophene over Pd/γ-Al2O3[J].J Catal,2005,235(1):229-240.
[5]SUN Y,PRINS R.Hydrodesulfurization of 4,6-dimethyldibenzothiophene over noble metals supported on mesoporous zeolites[J].Angew Chem Int Ed,2008,47(44):8478-8481.
[6]WANG L Y,WANG A J,LI X,et al.Highly acidic mesoporous aluminosilicates prepared from preformed HY zeolite in Na2SiO3alkaline buffer system[J].J Mater Chem,2010,20(11):2232-2239.
[7]WANG L Y,WANG A J,LI X,et al.A Novel Synthesis Method for Hydrothermal Stable Well-Ordered Mesoporous MCM-41From Preformed Zeolite ZSM-5[C]//Beijing,15th International Zeolite Conference,2008.
[8]王林英,王安杰,李翔,等 .以沸石为母体用降解法合成介孔硅铝分子筛[C]//第15届全国分子筛学术大会论文集,2009:608-609.
[9]QIAN W,ISHIHARA A,OGAWA S,et al.Study of hydrodesulfurization by the use of35S-labeled dibenzothiophene 1Hydrodesulfurization mechanism on sulfided Mo/Al2O3[J].J Phys Chem,1994,98(3):907-911.
[10]WANG A,KABE T.Fine-tuning of pore size of MCM-41 by adjusting the initial pH of the synthesis mixture[J].Chem Commun,1999,20:2067-2068.
[11]REN J,WANG A,LI X,et al.Hydrodesulfurization of dibenzothiophene catalyzed by Ni-Mo sulfides supported on a mixture of MCM-41and HY zeolite[J].Applied Catalysis A,2008,344(1-2):175-182.
[12]REDDY K M,SONG C S.Synthesis of mesoporous molecular sieves:Influence of aluminum source on Al incorporation in MCM-41[J].Catal Lett,1996,36(1-2):103-109.
[13]CHAKRABORTY B,VISWANATHAN B.Surface acidity of MCM-41by in situ IR studies of pyridine adsorption[J].Catal Today,1999,49(1-3):253-260.
[14]ZOU W,GONZALEZ R D,LOPEZ T,et al.The effect of precursor structure on the preparation of Pt/SiO2catalysts by the sol-gel method[J].Materials Letters,1995,24(1-3):35-39.
[15]CONŢESCU C,VASS M I.The effect of pH on the adsorption of palladium(Ⅱ)complexes on alumina[J].Applied Catalysis,1987,33(2):259-271.
[16]QIAN E W,OTANI K,LI L,et al.Hydrodesulfurization and hydrogenation reactions on noble metal catalysts PartⅡEffect of partial pressure of hydrogen sulfide on sulfur behavior on alumina-supported platinum and palladium catalysts[J].J Catal,2004,221(2):294-301.
[17]BARRIO V L,ARIAS P L,CAMBRA J F,et al.Hydrodesulfurization and hydrogenation of model compounds on silica-alumina supported bimetallic systems[J].Fuel,2003,82(5):501-509.
[18]BALDOVINO-MEDRANO V G,ELOY P,GAIGNEAUX E M,et al.Factors controlling the development of the HYD route of desulfurization of DBT overγ-alumina supported Pt and Pd catalysts[J].Catal Today,2010,150(3-4):186-195.
[19]SACHTLER W M H.Metal clusters in zeolites:An intriguing class of catalysts[J].Acc Chem Res,1993,26(7):383-387.
[20]SHEU L L,KNÖZINGER H,SACHTLER W M H.Palladium carbonyl clusters entrapped in NaY zeolite cages:Ligand dissociation and cluster-wall interactions[J].J Am Chem Soc,1989,111(21):8125-8131.
[21]YASUDA H,SATO T,YOSHIMURA Y.Influence of the acidity of USY zeolite on the sulfur tolerance of Pd-Pt catalysts for aromatic hydrogenation[J].Catal Today,1999,50(1):63-71.
[22]AMBS W J,MITCHELL J M M.Hydrogen spillover on platinum alumina,effect of water[J].J Catal,1983,82(1):226-229.
[23]MILLER J T,MEYERS B L,MODICA F S,et al.Hydrogen temperature-programmed desorption(H2TPD)of supported platinum catalysts[J].J Catal,1993,143(2):395-408.
[24]CONNER W C J,FALCONER J L.Spillover in heterogeneous catalysis[J].Chem Rev,1995,95(3):759-788.
[25]ZHANG H,MENG X,LI Y,et al.MCM-41 overgrown on Y composite zeolite as support of Pd-Pt catalyst for hydrogenation of polyaromatic compounds[J].Ind Eng Chem Res,2007,46(12):4186-4192.
[26]SUGIOKA M,ANDALALUNA L,MORISHITA S,et al.Noble metals supported on mesoporous silicate FSM-16 as new hydrodesulfurization catalyst[J].Catal Today,1997,39(1-2):61-67.
[27]GALLEZOT P.The state and catalytic properties of platinum and palladium in faujasite-type zeolites[J].Catal Rev Sci Eng,1979,20(1):121-154.
[28]STANKHEEV A Y,KUSTOV L M.Effects of the support on the morphology and electronic properties of supported metal clusters:Modern concepts and progress in 1990s[J].Appl Catal A,1999,188(1-2):3-35.
[29]GUCZI L,KONYA Z,KOPPANY Z,et al.Influence of pretreatment conditions on acidity of cobalt-based bimetallic systems in NaY zeolite[J].Catal Lett,1997,44(1-2):7-10.
猜你喜欢
分子催化(2022年1期)2022-11-02
中国特种设备安全(2021年5期)2021-11-06
贵金属(2021年1期)2021-07-26
贵金属(2021年1期)2021-07-26
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26
环境科技(2017年6期)2018-01-17
当代化工研究(2016年2期)2016-03-20
设备管理与维修(2016年6期)2016-03-16
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
山东工业技术(2014年19期)2014-08-15
