
金纳米棒的光学性质研究进展
2012-12-11 09:11柯善林阚彩侠朱杰君
物理化学学报 2012年6期
柯善林 阚彩侠,* 莫 博 从 博 朱杰君
(1南京航空航天大学理学院应用物理系,南京211106;2南京大学物理系,固体微结构物理国家重点实验室,南京210093)
金纳米棒的光学性质研究进展
柯善林1阚彩侠1,*莫 博1从 博1朱杰君2
(1南京航空航天大学理学院应用物理系,南京211106;2南京大学物理系,固体微结构物理国家重点实验室,南京210093)
金纳米棒在紫外-可见-近红外(UV-Vis-NIR)波段具有独特的可调节表面等离子体共振(SPR)光学特性,其良好的稳定性、低生物毒性、亮丽的色彩和在催化、信息存储、生物医学等领域广阔的应用前景受到相关研究领域的广泛关注.结合已有的研究基础,本文主要综述了金纳米棒光学性质的研究进展,包括表面等离子体共振、局域场增强效应、共振耦合效应及荧光特性,并对金纳米棒的应用做了展望.
金纳米棒;表面等离子共振;局域场增强;等离子体共振耦合;荧光
1 引言
现代技术的发展与变革在很大程度上依赖于现有的材料及新材料的产生:信息与数据存储要求材料介质能满足更大的存储密度和更高的传输速度;能源要求新材料能将太阳辐射有效地转换成电能等.在纳米材料的研究热潮中,贵金属(尤其是Au和Ag)纳米材料独特的光、电、催化等特性及其在新能源研究、光电信息存储、生物医疗等领域的应用受到众多研究领域的广泛关注.1-7在基础研究和应用探索的驱动下,研究人员发展并改进了多种贵金属纳米结构的制备策略.8-11近十年来,在Au纳米材料研究方面取得了长足的进步,人们研究了它们的可调制光吸收特性、催化活性及局域场增强效应与形貌和结构的相关性.12-142009年,研究人员利用Au纳米颗粒的局域场效应,研究了800 nm附近的双光子激发下Au纳米颗粒的发光性质,15发现了Au、Ag纳米颗粒与光学材料(如Nd3+掺杂的玻璃,SiO2、TiO2纳米材料等)的复合能够增强的发光效率.16,17最近,人们发现当金属(Au、Ag、Al等)纳米粒子相互靠近形成二聚体、三聚体、一维链及二维阵列时,纳米粒子的表面等离子体共振会出现耦合效应.这种耦合效应在粒子的局域产生强烈电磁场,这种增强效应能够有效地提高分子的荧光产生信号、18-20分子的拉曼散射信号、21-23双光子或多光子发光、24-26二次谐波增强27,28等非线性过程.
结合已有的实验与理论研究基础,本文综述了金纳米棒光学性质的研究进展,并对金纳米棒的未来应用做了展望.
2 金纳米棒的光学性质
许多金属表面(如碱金属Al、Mg和贵金属Au、Ag等)的自由电子都可形象地看作电子气,电子气的集体激发称作等离子体,它是金属表面自由电子同入射光子相互耦合形成的非辐射电磁模式.不同金属等离子体的频率决定了各自的光学性质,当光的频率低于金属的等离子体频率,光会被反射回来.绝大多数金属的等离子体子频率在紫外区域,所以我们看到多数金属的颜色是可见光复合而成的白色.由于Au(Ag及Cu)的电子结构比较特殊,带间跃迁发生在可见光波段,对一些特定波长的光有很强的吸收,所以它们看起来有独特的颜色.
在纳米材料光学性质研究中,金属纳米颗粒优异的光学性质源于其表面等离子体共振(SPR).紫外、可见和近红外区域的光入射到金属和介质的界面时,当满足所有的边界条件,将会激发金属颗粒表面价电子的集体振荡,即SPR.由于共振使电子吸收了入射光的能量,从而使反射光在一定角度内大大减弱.Au纳米结构在可见至近红外较宽波段表现出体相材料中所观察不到的强吸收带,这也是我们经常会看到不同形状和尺寸Au纳米颗粒胶体溶液呈现五颜六色的原因所在.因此,金属纳米颗粒的重要光学特性是SPR频率与颗粒的形状、尺寸、组分、环境的介电常数有密切的关系.29-32
2.1 等离子体、等离子体共振的理论模拟
相对于理想金属体材料的传导型等离子体,纳米材料具有较大的比表面,且表面较为粗糙,它对应着另一类束缚(或局域)模式,即局域表面等离子体(LSP).LSP是被局域在不同形貌的曲面上的一种非传播模式.当光入射到金属纳米颗粒表面时,如果入射光子频率与金属传导电子的整体振动频率相匹配,纳米颗粒会对光子能量产生很强的吸收作用,就会发生局域表面等离子体共振(LSPR)现象,331902年,Wood34在光学实验中首次发现了SPR现象.1908年,为解释任意尺寸球形颗粒的光学性质,德国物理学家Mie35通过求解球极坐标系中Maxwell方程(利用场在颗粒表面边界条件)提出Mie理论.在Mie理论基础上,其他科学家发展出非球形颗粒光学性质的理论或方法,如线性颗粒的Gans方程、异质球形复合颗粒(芯-壳结构)循环解决方式及截角三棱柱的离散偶极近似(DDA)方法.36-39
球形纳米金颗粒由于结构上的高度对称性,等离子振动也是各向同性的,表现为单一的SPR峰.与球形的纳米金颗粒相比,棒状Au纳米颗粒由于结构的各相异性,导致各个方向上电子的极化程度不同,振动模式如图1所示,由此产生了两个表面等离子体共振模式.随着长径比的增加,两个表面等离子体共振吸收峰的频率(或波长)分离也增加.高频率(短波长)共振峰由垂直于棒轴向的电子共振产生,称之为横向SPR吸收,位于510-530 nm范围;另一个在较大波长范围内移动的共振峰由沿着纳米棒轴向的电子共振产生,称为纵向SPR吸收.40随着纵横比的变化,横向SPR(SPRT)吸收峰位置变化较小,而纵向SPR(SPRL)峰的位置可以在可见-近红外较宽波段内移动.因此,不同Au纳米棒胶体溶液可以呈现出蓝色、绿色、褐色等不同颜色.
对于任意形状和尺寸的颗粒,T-矩阵(麦克斯韦方程的线性与场中颗粒的边界条件保证了散射与入射场的线性关系,这两套系数间的线性形变称为T-矩阵)是较普遍的方法.以下是经过简化后适用于计算椭球状颗粒吸收截面的Gans方程.41

图1 棒状金属纳米粒子的两种等离子体振荡示意图Fig.1 Schematic of two plasmon resonances of metallic nanorods(a)SPRTof nanorod,(b)SPRLof nanorod;T:transverse,L:longitudinal

其中,εm为介质相对介电常数,ε=ε1+iε2为颗粒的介电函数,pj(j=a,b,c;a代表长度,b=c代表宽度)表示消偏振因素,e表示椭圆度,V=4abcπ/3.
根据金属团簇光学性质的理论模型和Gans方程,利用Au的介电函数,42可以计算出具有不同纵横比的Au纳米棒的光吸收谱(不考虑尺寸分布及介质介电函数变化的影响),如图2所示.从理论谱中可以看出:在可见光和近红外光区分别出现较弱的SPRT与较强的SPRL两个吸收峰,其中SPRL随着纵横比的减小线性蓝移,而SPRT随着纵横比的减小在很小的范围内红移.
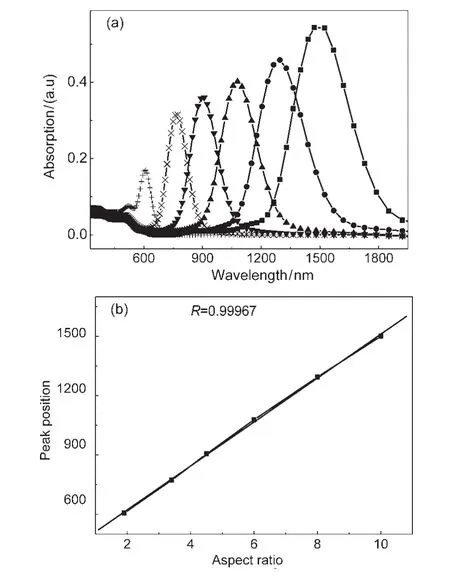
图2 (a)Au纳米棒的理论光吸收谱;(b)SPRL的峰位与纵横比的线性关系40Fig.2 (a)Calculated absorption spectra of theAu nanorod;(b)linear relation between the longitudinal plasmon band position and aspect ratio40In Fig.(a),different aspect ratio:10,8,6,4.5,3.5,1.8 (from right to left)
随着纳米材料制备技术的发展与完善,不同形貌的纳米颗粒在实验中成功合成.在等离子体共振理论模拟上,Yang等43提出了DDA理论.DDA理论将所研究的纳米粒子视为有N个点偶极子构成的立方阵列,由每个点偶极子的极化率张量积分而得出颗粒的吸收截面.DDA理论已逐渐发展成为表征任意形状金属纳米粒子的吸收、散射和消光等光学性质的非常重要的手段.
目前,较常用的另一种模拟方法是时域有限差分法(FDTD).FDTD方法是Yee44在1966年提出的.采用Yee元胞的方法,在空间、时间上对电场强度和磁场强度进行离散.如果知道材料的介质参数及介电常数与波长(或频率)之间的关系,采用数值计算的方法就可以在时间轴上步步递推地求解空间电磁场分布.为了便于分析光吸收及电子振荡过程, FDTD可将电磁场随时间的演化关系用不同的颜色显示,以表示局部电磁场的强度.FDTD在多个领域获得广泛应用,如:辐射天线分析、微波器件和导行波结构的研究、电磁脉冲的传播和散射、周期性结构分析、微光学元器件中光的传播和衍射特性、分析环境和结构对元器件和系统电磁参数及性能的影响及电子封装、电磁兼容分析等.
2.2 表面等离子共振
当可见光照射在Au纳米粒子表面时,和共振波长相同的光被吸收并诱导表面电子集体共振.由于Au纳米粒子的LSPR与其形状、大小、表面介电常数等密切相关,所以大小、形状、聚集程度以及所处的局部环境不同的Au纳米粒子具有不同的LSPR(包括峰的数目、峰形、峰位、峰宽),其胶体溶液便可以呈现出各种不同的颜色.45比如Au纳米粒子从球形径向生长成纳米棒时,粒子会呈现与纳米棒径向比对应的红色、橙色、绿色、蓝色等各种颜色.另外,小粒径的球形Au胶体溶液伴随着聚集的发生,其LSPR吸收带会发生红移或展宽,相应的颜色从红色变至蓝色或紫色.如图3所示,这些肉眼可见的颜色反映了传导带电子(等离子体)在适当波长的光照射下的相干振荡.而由等离子体共振引起对光的强烈吸收和散射,构成了Au纳米粒子在生物传感和成像研究中的基础.46
根据实验结果,我们利用FDTD方法模拟了实验中合成的Au纳米棒的理论光吸收谱,如图4(a) (选取折射率为1.33,图中数据1,2,3,4,5对应纵横比(L/D)分别为2,3,4,5,6)所示.从模拟结果可以看到横向峰的位置大约在530 nm,随着纵横比的增加,横向峰有一定的蓝移,移动很小,峰值有一定的增加,而纵向峰的位置变化较大,出现较大的红移,可以移动到红外区,且峰的强度有很大的增加.这是由于棒状的纳米结构在入射光的激发下,正负电子在两个极化方向上出现了分离,从而形成了横向和纵向的等离子体振荡,导致了两个共振峰.通过对比同一纵横比的戴帽(capping ends,即棒两端有五重孪晶面)和不戴帽(五棱柱)的Au纳米棒的光学吸收谱,发现戴帽的纳米棒纵向峰发生蓝移,且吸收峰强度有所减弱.这是由于帽形成了尖端,电荷分布发生了改变,其振荡方式和强度也发生改变,可以有效控制峰的位置.图4(b)是我们利用FDTD模拟的截面为八边形的单晶Au纳米棒胶体的光谱图,插图为实验所得Au纳米棒的TEM结果和相应样品的光吸收谱(纳米棒的纵横比约4.5).47对比实验光谱和理论光图,我们能发现光谱图中SPR变化基本趋于一致,但SPR峰的峰位和形状略有差异.这是由于实验光谱图是大量纵横比不均一的纳米棒和少量副产物(如颗粒)吸收的叠加结果,且环境折射率变化(表面活性剂引起)和棒的横截面形状(对称性)对SPR也产生明显的影响.

图3 不同粒径的金纳米球与金纳米棒的胶体溶液和相应胶体的透射电子显微镜(TEM)图46Fig.3 Photographs of aqueous solutions and TEM images ofAu nanospheres andAu nanorods as a function of increasing dimensions46scale bar=100 nm;(A)Au nanospheres,(B)Au nanorods
为了研究环境折射率对光学吸收的影响,我们模拟了分散在不同介质(折射率为1.0、1.1、1.2、1.3、1.4、1.5)中纵横比为5的五重孪晶Au纳米棒的光学吸收谱,结果如图4(c)所示.可以看出:随着折射率的增加,横向SPR吸收峰变得越来越明显,并有较小的红移;纵向SPR吸收峰出现了较大红移.这种SPR峰位与周围介质的高度敏感性可以用来检测催化反应等过程中介质的变化.14,48
另外,贵金属纳米颗粒的SPR特性还受到多种因素的影响.例如,Manikandan等49认为,当金属纳米颗粒的尺寸较大时,颗粒的高阶振动模式(四极子和八极子)不能被忽略,导致吸收光谱发生蓝移. Marinakos等50证实了当Au纳米棒表面发生键合反应而改变其周围环境的介电性质时,其SPR峰也会发生移动.通常水溶液中合成的Au纳米棒取向随机分布,实验光谱表现出各个取向Au纳米棒SPR的平均效果.为解决这些问题,Li等51采用了一种薄膜加热和拉伸方法,成功将大量Au纳米棒按照拉伸方向排列起来,使得宏观上线偏光能够有选择地同时激发所有Au纳米棒的纵向或横向SPR吸收.进一步研究发现,Au纳米棒的定向排列能够还原单个Au纳米棒与极化相关的非线性特性,提高薄膜材料的非线性各向异向指数.同时,样品的非线性吸收系数被提高近1-2个数量级,这一结果与颗粒间耦合效应密切相关(见2.4节).
2.3 局域场增强效应
金属纳米颗粒在可见光和近红外光的照射下,等离子体共振引起的颗粒表面数纳米范围内强烈的局部电场,可比入射电场增强几个数量级,金属的这种表面局域场增强,可以极大地提高表面增强拉曼散射、高次谐波产生、双光子发光等非线性过程的转换效率,因而成为国际上物理、化学、材料科学和纳米科技领域众多研究人员长期普遍关注的话题.其中,金属纳米颗粒的表面增强拉曼散射能够大大增强分子光谱的信号强度,直至实现单分子光谱探测,52-58因此得到了广泛而深入的探索和研究.

图4 FDTD模拟的Au纳米棒不同截面的光吸收谱Fig.4 Simulated absorption spectra using finite-difference time-domain(FDTD)forAu nanorods with different cross sections(a)Au nanorods with five-fold twin cross section.solid curve:capped nanorods,the vertex distance is 10 nm,the height of the cap is 6 nm. dashed curve:nanorods without caps;(b)Au nanorods with octahedron cross section.Insets show the absorption spectra of aqueous solutions and TEM image of theAu nanorods;47(c)capped nanorods are immersed in different media with aspect ratio of 5.
金属纳米颗粒的局域电磁场增强效应,其物理根源是纳米颗粒表面的自由电子在电磁场的驱动下,在颗粒的特定部位发生强烈的电荷集聚和振荡效应,即在颗粒的近场区域产生强烈的电磁场,该部位称为“热点”(hot spots).为实现单分子检测的目标,必须要求局域电磁场有显著的增强.在过去20年内,学术界提出了几种方案,包括银纳米颗粒的溶胶聚合体(增强因子G=1014-1015),银纳米颗粒二聚体(G=1010
-1011),针尖增强方案(G=1010-1011),以及具有尖锐边角的纳米颗粒(G<1010)等.前两种方案结构复杂、机械稳定性差,而且热点区域很小,所占空间比例很低,不利于信号的观测.第三种方案需要使用复杂的装备,而第四种方案增强因子有限.为解决以上的困难,实现单分子检测的根本目标,理想的方案是设计、合成具有足够大局域场增强因子的单个金属纳米颗粒和利用等离子体的耦合效应提高场强.
由于金属纳米颗粒在可见光波段都存在宽带吸收损耗,所以一般金属纳米颗粒的局域电磁场增强因子较小.为了提高颗粒表面电场增强因子,就要将等离子体波的振荡局限在一个很小的区域,即产生局域的电场增强效应.利用经典电磁理论,对Au纳米颗粒局域场增强效应数值分析结果表明,在550-900 nm波段范围内,Au纳米颗粒均具有较强的局域场增强效应,这为深入研究不同Au纳米颗粒的非线性光学性质提供了重要的理论参考价值.
Au纳米颗粒局域电磁场增强效应一个最重要的应用领域是表面增强拉曼光谱(SERS).假设入射光电场振幅为1,局域电磁场振幅大小为│E│,则局域场的强度增强因子为│E│2,而SERS信号的增强因子为G=│E│4.Au纳米颗粒表面强烈的电场能把吸附分子的拉曼信号增强几个数量级,从而可以缩短拉曼信号收集时间,提高分析灵敏度.拉曼光谱在物质分子结构及探测分析物质化学信息研究中发挥很大作用.目前,具备拉曼增强效应的主要材料除了Au纳米颗粒外,在Ag、Cu、Pt、合金及半导体52,59,60等基底上也发现具有表面增强效应.
拉曼增强主要有两种机理,即化学增强机理61,62和电磁场增强机理.63,64化学增强信号的强弱决定于吸附分子的本身特性,化学增强是由于电子在分子和颗粒表面间转移而引起的分子极化的加强.62,65不同分子吸附在同一衬底上,增强效果也不相同.即使是同种分子,和不同金属纳米颗粒表面(尤其“热点”)结合也不同,增强效果也不相同.电磁场增强包括SPR、避雷针效应等.64SERS的电磁场增强机理通常用SPR解释,即金属颗粒表面的粗糙度提供了光与表面等离子体耦合的必要条件.当粗糙的金属基体表面受到光照射时,金属表面的电子被激发到高的能级,与光的电场耦合并产生共振,从而使金属表面的电场增强,产生表面增强拉曼信号.理论计算表明:与球形颗粒相比,Au纳米棒的末端极化电荷密度较大,电场较强,所以Au纳米棒更适合作为拉曼增强的衬底.66-68
Li和Xia69在计算中考虑的复合纳米颗粒是单个空心的Au立方盒状颗粒,颗粒中央的区域填充有增益介质,其光学性质由复数折射率n-ikcore来表征.Au颗粒的外边长为40 nm,内边长为32 nm,侧壁的厚度为4 nm.纳米颗粒的周围环境为水,折射率为1.33,增益媒质的折射率为1.33-ikcore,增益系数kcore可以变化,相应地该复合纳米颗粒的散射、吸收和消光光谱也将发生改变.计算表明,在共振波长779.2 nm处,当复合增益系统处于共振,即在kcore=0.143处金纳米颗粒的最大场增强因子为4.2× 108,相应的SERS增强因子十分巨大,为1.8×1017,而纳米颗粒外表面的局域场最大增强因子为2.4×108,相应的SERS增强因子也达到6×1016,已经超过了单分子检测的水平.计算还发现,场增强不仅仅局限于有限的几个“热点”,而是弥散在整个纳米颗粒的立体空间和内外表面,表面的平均场增强因子达到1×108,相应的SERS增强因子达到1×1016.
与单个Au纳米棒颗粒的增强效果相比,端-端相对的Au纳米棒二聚体及多聚体在电场激发时会引起电荷的重新分布,并在其缝隙处发生近场耦合效应,这种耦合效应可以大幅度提高局域电磁场增强因子和发光材料的发光效率.接下来详细评述Au纳米棒的耦合效应和荧光效应.
2.4 等离子体共振耦合
SPR耦合即当两个纳米颗粒靠近,一个颗粒的电子振动将引起另外颗粒表面电子的振动,它们的SPR性质相互影响,两个颗粒的SPR就会发生耦合,导致SPR的光学性质发生变化,而成为近几年的研究热点.15,70-75这种耦合(尤其是尖-尖耦合与端-端耦合),如图5所示,产生的局域电场增强效应通常比单个颗粒的表面电场增强效应大得多.28,76,77.然而,纳米颗粒间的表面等离子体共振耦合效应受到金属颗粒间距的影响很大(随着距离的增大,耦合迅速衰减).目前,制备这种结构主要是用离子刻蚀法.但受到模板尺寸的影响,离子刻蚀法能将颗粒间距减小到约10 nm.因此,要实现等离子体共振的耦合,人们需要借助于配位体的物理作用、化学交联、两相法、模板法、衬底表面原位生长等方法,将金属纳米颗粒在液相中或平面衬底上拉近颗粒间距(仅有一个至数个分子大小).有关纳米粒子相互靠近形成同型或异型的二聚体、三聚体、一维链及二维阵列,纳米粒子的SPR引起的耦合效应有诸多的报道,如纳米球、72,78-81棒、82-85圆盘、86,87环、88,89管、90,91壳层、92-97多面体98-100等规则形貌及无规则形貌101-105的纳米结构.但是粒子间距小于2 nm时,在实验上仍面临着挑战.

图5 FDTD模拟的Au纳米蝴蝶结不同区域的增强效应54Fig.5 FDTD simulation of local field enhancement on the tip ofAu nanostructures54
对于Au纳米棒的二聚体耦合,我们利用FDTD方法模拟了两个纵横比(r)为3(20 nm×60 nm)的五重孪晶金纳米棒按照不同耦合方式的光谱(折射率为1.0),如图6所示.对于双棒按照端-端相对的结构,光的极化方向沿着棒的轴向(即光沿X方向垂直棒照射),将产生纵向的共振模式.然而从图6(a)中可以看出:在约650和1500 nm波段出现两个较明显的共振峰,即Au纳米棒的纵向SPR峰和双棒耦合激发的位于较长波长(1500 nm)的振荡模式.随着间隙距离的增加,Au纳米棒的纵向SPR峰发生红移(见插图,这与已有的文献83报道有矛盾,值得注意的是在波长较长的范围内出现了新的耦合峰,这是文献中所没有的),这是由于在两棒尖端耦合强度(或局域电场强度)随距离增大而减小,共振能量降低,从而导致红移.耦合激发的新吸收峰是两个纵向共振模式相互作用的结果,另外由于对称性破坏,也可以产生额外的共振模式.当间隙距离为5 nm时,耦合效应消失,1500 nm处的耦合峰也随之消失.图6(b)是纵横比为3和6的纳米棒及耦合双棒(r=3,间距为1 nm)的吸收谱.可以看出:耦合双棒的吸收不能等效为长棒(r=6)的吸收,也不是两个单棒(r=3)的简单相加,这正是双棒耦合作用的结果.图6 (c)是边-边相对双棒的模拟结果.

图6 FDTD模拟五重孪晶Au纳米棒二聚体按照不同耦合方式的光谱图Fig.6 Simulated absorption spectra using FDTD for five-fold twinedAu nanorods dimer arranged in different configurations(a)absorption spectra as a function of inter-particle distance for twoAu nanorods(dimensions:20 nm×60 nm,i.e.,aspect ratio r=3)aligned end-to-end along Z axis;(b)absorption spectra for monomer(solid line:r=3 and dash line:r=6)and a pair ofAu nanorods(dot line,r=3,spacing=1 nm)aligned end-to-end along Z axis;(c)absorption spectra as a function of inter-particle distance for a pair ofAu nanorods(r=3)aligned side-by-side along the Z axis.inset:absorption spectra for monomer(solid line,r=3)and a pair ofAu nanorods(dash line,spacing=0.1 nm)aligned side by side.For all calculations,rods are immersed in a homogeneous medium with a refractive index of 1,the polarization is along the Z axis.

图7 FDTD模拟Au纳米棒单体和二聚体按照不同耦合方式的场增强分布图Fig.7 Simulated electric field enhancement using FDTD for a singleAu nanorod and dimersAu nanorods dimer aligned end to end with spacing of 0.1 nm(a),1 nm(b),and 5 nm(c);Au nanorod monomer with aspect ratio of 3(d)and 6(e);Au nanorods dimer aligned side-by-side with spacing of 1 nm(f)

图8 (A)Au纳米棒二聚体不同耦合光散射谱;(B)不同构型二聚体耦合示意图;(C)用DDA方法得到的耦合金纳米棒二聚体表面电荷密度83Fig.8 (A)Scattering spectra of variousAu nanorod dimers;(B)plasmon hybridization schemes for dimers in different geometric arrangements;(C)surface charge density of coupling gold nanorods dimer calculated using discrete dipole approximation(DDA)83Inset in Fig.(A):electron micrographs of the same dimers showing the different configurations,scale bar=100 nm; (B)red×means impossible configurations;(C)interparticle separations are all 1.5 nm.
图7为我们用FDTD模拟Au纳米棒单体和二聚体按照不同耦合方式的场增强分布图(模拟时,采用相互垂直的双光源,场增强的数量级,即彩色标尺的数量级,只具有相对意义.一般,文献中采用FDTD方法模拟时,场增强没有数量级,只有颜色标尺).从图7(a,b)中可以看出:端-端相对的双棒间距为0.1 nm时,两棒耦合的电场增强效应比间距为1-4 nm的要大一个数量级,说明间距非常小时,其耦合非常大.相比于单棒的增强效应,双棒端-端间距为0.1 nm时电场要增大4个数量级,即双棒端-端间距为0.1 nm时,拉曼散射增强因子增大8个数量级.说明颗粒间耦合可以大大改善其作为拉曼散射基底的应用.双棒耦合不同于两个单棒的简单叠加,如图7(d,e)所示,纵横比为6的单个纳米棒的增强效应只比纵横比为3的单棒大一个数量级.
Funston等83用DDA方法模拟了金纳米棒二聚体四种构型的耦合,如图8所示.图8A中(a,b)是端-端相对的纳米棒二聚体,当光极化方向平行于棒(纵向的共振模式)时,得出的结论和我们用FDTD方法模拟的结果基本吻合,在约820 nm处出现新的共振峰.当光极化方向垂直于棒时,没有观察到共振峰的明显移动,这种情况下耦合较弱.图8A中(c,d)是纳米棒边-边相对放置,计算模拟表明,当光极化方向平行于粒子间距(即垂直于棒,横向的共振模式)时,随着棒间距的减小,纵向峰整体略微蓝移,因为横向等离子体相互作用较弱,共振吸收峰主要受控于纵向等离子体共振.从物理学的角度上看,端-端相对与边-边相对两种情况类似对应于两个弹簧振子的串联和并联,主要考虑纵向模式,当光极化平行于二聚体长轴时,端-端相对棒相对两端电荷相互吸引导致棒中的电荷共振恢复力减弱;当光极化垂直于二聚体长轴时,边-边相对每个棒两端电荷相互吸引,导致棒中的电荷共振恢复力增强.106,107图8A中(e,f)是金纳米棒二聚体的T与L两种耦合情形,对于T构型,是一棒的横向模式和另一棒的纵向模式相互耦合,当光极化方向平行于粒子间距时,导致了T构型主干以纵向SPR模式;相反,当光极化方向垂直于粒子间距时,导致T构型顶端纵向SPR模式.对于L构型,两粒子共振耦合均为纵向模式而不是纵向与横向的耦合,当光极化方向平行于粒子间距时导致低能量带的激发,两个纵向共振带相互吸引,同相位振荡,产生了相对立的偶极子.当光极化方向垂直于粒子间距时,两个纵向共振带异相位振荡,两偶极子相互排斥.图8B是各种构型纳米棒的耦合图,其中红×表示该耦合的电场不能实现.从图8C中可以看到各种构型耦合下纵向激发的电荷分布情况.
目前,虽然已有大量的文献报道同型二聚体纳米粒子的耦合,以及把互相耦合的两个粒子看作一个整体,着重讨论整个体系的共振行为与体系中颗粒的形貌、间距、数目、相对位置以及环境介电常数的关系,且只局限于理论模拟,但对于异型二聚体纳米粒子的耦合很少关注,最近鲜有报道.108-111
Yao等112用FDTD方法模拟了具有不同性质的纳米颗粒与Au纳米棒的耦合,探讨了引入颗粒的等离子体共振频率(见图9)、体积等因素对Au纳米棒在正入射条件下,诱导出无法激发的偶极纵向等离子体模式(禁戒模式)的影响.研究发现:当外来颗粒的等离子体振荡频率与Au棒的禁戒模式频率一致或者外来颗粒体积足够大时,可以使Au纳米棒的禁戒模式被有效地激发.这一结果可以很好地解释最近报道的用近场光学显微镜研究Au纳米棒等离子体振荡的实验结果.他们认为外来颗粒表面电荷积聚造成强的电场对称性破缺是诱发Au棒禁戒模式的原因.外来颗粒的等离子体共振越强、体积越大,电荷的积聚就越多,通过耦合诱发Au棒禁戒模式的能力就越强,而相对于金属颗粒中的自由电荷,介质颗粒中的极化电荷产生的电场较弱,不能有效诱发禁戒模式.
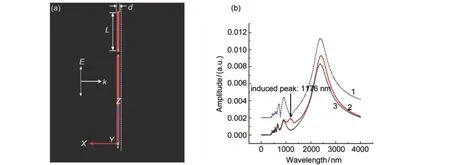
图9 (a)共振、非共振短棒与长棒耦合的示意图;(b)共振(line 2)、非共振(line 1)短棒与长棒耦合时长棒的光谱及长棒单独存在时的光谱(line 3)112Fig.9 (a)Configuration for simulating the coupling of the longAu nanorod with the resonant and non-resonant short rod; (b)optical spectra of the longAu nanorod coupled with resonant(line 2)and non-resonant(line 1)short rods,and optical spectrum of isolate longAu nanorod(line 3)112(a)resonant rod:m=1 mode at 1268 nm,non-resonant rod:m=1 mode at 723 nm.The separations between the nanorods are 10 nm in both cases. The arrow indicates an additional peak induced by the resonant short rod.
此外,如果把Au纳米棒排列成三聚体、一维链、二维阵列,粒子间的SPR会发生耦合作用,产生一系列集体效应.Au纳米棒末端产生巨大的电磁场增强,可用于提高拉曼散射,66-68强烈的近场相互作用能够把电磁场的能量从一个Au纳米棒沿着链的方向转移到另一临近的纳米棒,可用于传输光信号的波导,45,113二维阵列的结构能够在远场成像,突破近场传输图像的限制.114
Ming等115报道了一种基于可控共振耦合的表面等离子体开关元件.这样一个开关由单个Au纳米棒和其周围的光致变色分子组成,大小不到100 nm,Au纳米棒和分子都被封装在一层SiO2薄膜中.而它的开关属性则是由紫外光来激发,由暗场散射技术来监测.操纵这样单个表面等离子体开关所需要的触发功率和能量只有大约13 pW和39 pJ.这种光控等离子体开关可以作为纳米光子线路中的一个开关元件,从而能够与微电子元件很好的耦合,解决它们之间的尺寸匹配问题.
3 金纳米棒的应用
Au纳米棒呈现强的光散射和吸收特性及良好的荧光特性,特别是其可调节的SPRL峰,使其更适合在生物医学、生化标记及成像分析中做光学探针.接下来我们简要讨论Au纳米棒荧光特性的机理,并给出其在一些重要领域中应用的例子.
3.1 荧光特性的机理
Au纳米棒的荧光特性已经有许多实验和理论计算的报道,116-118宏观贵金属由于电子带间跃迁,本身具有荧光特性,不过荧光极其微弱.研究表明,与宏观体材料相比,Au纳米粒子具有较强的荧光效应,尤其是纳米棒,团簇等.119,120
Zhu等121测定了纵横比为2.5的Au纳米棒的荧光光谱,发射峰位于370和670 nm;孙桂敏等122制备了纵横比为8的Au纳米棒,在480 nm波长激发下,在560和707 nm波长处有两个荧光发射峰;Li等118发现相当长的Au纳米棒(纵横比大于13)在690 nm波长激发下,有743 nm(较强)和793 nm(较弱)两个特征荧光发射带.由于与SPR峰产生的局域场耦合作用,Au纳米棒的吸收及发光效率大大提高,长Au纳米棒比短Au纳米棒荧光效率更高.采用荧光发射和双光子诱导光致发光两种技术可以检测到低荧光量子产率的短Au纳米棒的增强荧光.15
Au纳米棒除本身具有荧光外,还可增强有机荧光染料分子、量子点等纳米粒子的荧光.我们知道Au纳米棒的LSPR会导致局域场增强效应,如果将Au纳米棒与有机荧光染料分子或量子点二者结合起来,它们的发光效应大大提高.表面增强的荧光效应根源在于SPR产生的电磁场.123,124荧光增强来自于激发增强和发射增强两部分.当纳米粒子等离子体共振波长接近分子发射波长时,发射增强得到提高,发射增强因子fem可表示为125,126
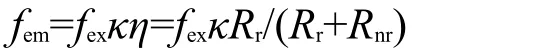
这里,fex是激发强度的增强因子,κ为光收集效率,η是量子产率,Rr和Rnr分别为辐射性衰变和非辐射性衰变率.激发增强因子fex和局域场增强成比例.
Ming及其合作者127采用Au纳米棒内核、可生物修饰的硅壳以及修饰在硅壳表面的生物有机染色分子构成的复合结构,证实了Au纳米棒的荧光增强因子取决于横向与纵向极化激发的平均电场强度增强之间的比例,并得出了荧光强度和激发极化角度有关.在横向极化激发下,局域场增强很小,当Au纳米棒的纵向等离子体波长和激发波长几乎相等时荧光增强达到最大.
Li等126用CdSe做为量子点,用正硅酸乙酯做稳定剂,利用静电作用将量子点和抗体Anti-CEA8联接到Au纳米棒-二氧化硅核壳结构上,当Au纳米棒的LSPR和量子点发射谱相匹配时,会导致量子点荧光淬灭.当两粒子距离靠近时,辐射性衰变率增加,而非辐射性衰变率降低,然而这两者对荧光增强而言都是负面的,适当的增加Au纳米棒壳层厚度,可使量子点荧光增强最优化.实验证实当量子点距Au纳米棒15 nm左右时,量子点发射增强达最大.在实验中测量的量子点荧光寿命和辐射性衰变率和原来的数值并没有多大变化,说明了荧光衰减率的变化可忽略,主要是由于激发增强导致荧光增强所致.
3.2 生物医学中的“荧光探针”
在生物科学领域中,活体生物组织的实时成像是人们一直追求的目标.但荧光成像技术面临着诸多难题,例如,细胞在可见光区的自发荧光对标记分子所发信号的掩盖,对所研究分子很难进行长期荧光标记观察等.这就迫切需要研制开发光稳定性好的近红外荧光探针.Au纳米棒的SPRL通过调整纵横比可以精确调控在600-1500 nm区域,而这范围(尤其是近红外范围)正是生物组织的最佳透过波段.Au纳米棒是一种理想的“双光子荧光”成像类型,能比常规的荧光影像提供更高的对比度和亮度.Au纳米棒作为一种荧光探针已被广泛应用在生物医学等领域.
Qian等128利用荧光和表面增强拉曼散射共同作用的Au纳米棒作为近红外探针,在活鼠的有机体获得了纯光学图像.Au纳米棒经3.3ʹ二乙基硫醛三碳菁化碘(DTTC)修饰,外面附着一层硫基聚乙醇(PEG-SH)作为稳定层.他们首次通过纯光学图像观察到静脉注射到活鼠有机体深层组织的Au纳米棒的分布和排泄.这种技术相比其他成像技术具有很高的对比度和深层的探测能力,将来可能会取代传统的放射性医学成像技术,在疾病诊断和临床医学有巨大的应用前景.
Durr等129用Au纳米棒作为造影剂,借助双光子发光成像技术来探测肿瘤细胞,在Au纳米棒上标记anti-EGFR(表面生长因子蛋白),分子特异性成像能深入75µm以下的组织,并且具有很高的信噪比.用760 nm的激光光源,从Au纳米棒标记的肿瘤细胞获得的双光子发光强度是无标记的肿瘤细胞自身发出荧光的3倍.此外,他们还发现Au纳米棒可以扩展双光子成像的能力,是一种非侵入式、三维成像的新方法.
孙桂敏及其合作者122利用静电作用将Au纳米棒非特异性标记到HepG2人肝癌细胞的表面, HepG2细胞在可见-近红外光区无任何吸收,而标记了Au纳米棒的HepG2人肝癌细胞其吸收光谱图出现了Au纳米棒的特征吸收峰.荧光实验进一步证明了Au纳米棒的作用,采用480 nm波长激发时, HepG2人肝癌细胞收集不到荧光信号,而当Au纳米棒标记细胞后,荧光光谱则显示出了Au纳米棒的特征荧光峰.此外,Au纳米棒标记的HepG2细胞在488 nm波长激发下,获得了绿色和红色两种颜色的荧光图像,而未标记组细胞则不呈现荧光.由于Au纳米棒的抗光漂白能力较强,用氙灯连续照射4 h后,其荧光强度仅下降2.4%,远低于传统有机染料.由此,Au纳米棒作为荧光探针,有望代替传统荧光染料应用于细胞的荧光标记.
最近,Wang等130将Au纳米棒和Fe3O4纳米粒子组合制备了一种新的荧光探针(图10).这种荧光探针能靶向识别SK-BR-3细胞,进行磁共振成像,同时Au纳米棒能吸收近红外光的能量并将其转化为热能,来杀死癌细胞达到治疗的目的.这种新型的纳米探针在癌症治疗方面有很广阔的应用前景.他们还将Fe3O4纳米颗粒定位组装到不同比率的Au纳米棒的两端和侧面,分别构筑成Fe3O4-Aurod-Fe3O4哑铃型和Fe3O4-Aurod珍珠项链型结构的纳米颗粒,这种材料同时具有磁性和光学可调性.131在制备过程中通过调节纳米棒的纵横比,将其SPR由可见区调到近红外区,这样不但可以检测多元目标物,且Au纳米棒经近红外光照射后,通过吸收近红外激光能量,能迅速升温“热死”细菌,还可利用Fe3O4纳米颗粒的超顺磁性达到分离目标物的目的.
3.3 生化标记及成像分析中的“光散射探针”
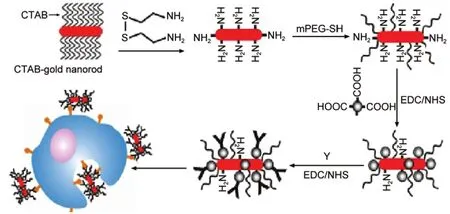
图10 Au纳米棒/Fe3O4纳米粒子生物探针的合成和靶向示意图131Fig.10 Schematic of synthesis and targeting ofAu nanorod/Fe3O4bioprobes131CTAB:cetyltrimethylammonium bromide,EDC/NHS:1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hychloride/N-hydroxysuccinimide,Y:herceptin
由于独特的LSPR光散射特性,Au纳米棒在白光的照射下可以散射出随其粒径、形态而变的特定颜色的光,而且散射光很强,抗体、DNA探针和其它的示踪物质能够很容易地连接到Au纳米棒上,不会改变它们的光散射性质,能达到单个有机荧光分子的荧光效率的好几个数量级倍.此外,Au纳米棒有较高的散射量子产率,不会被光漂白,非常适合长时间的观察和研究,相比半导体荧光量子点的生物毒性,生物相容性好,在细胞成像及活体检测方面更具有优势.
郭红燕等132将拉曼活性分子对巯基苯甲酸吸附于Au纳米棒表面,制备出SERS标记的纳米棒探针,该探针和蛋白抗体结合形成SERS标记抗体.通过SERS标记抗体、待测抗原和俘获抗体(固体基底上修饰的抗体,即俘获抗体)之间的免疫应答反应,将Au纳米棒探针组装到固体基底上,形成SERS标记抗体-抗原-俘获抗体“三明治”夹心复合体,通过纳米棒的SPR和激发光的耦合来优化SERS信号,单组分抗原的SERS免疫检测浓度范围高于1×10-8mg·mL-1.
He等133利用杂交后的双链DNA具有带负电荷的双电子层,而单链DNA不具有负电荷双电子层,对于CTAB包覆带正电荷Au纳米棒来说,可以通过静电吸引作用拉近Au纳米棒的距离,使等离子体共振散射信号显著增强的原理,建立了检测HIV-1相关DNA的方法.
Huang等134利用生物标记的Au纳米棒来识别癌变细胞.癌细胞在生长过程中会过量地表达EGFR,使用抗表面生长因子蛋白抗体标记的Au纳米棒与细胞孵育后,抗体修饰的Au纳米棒就会因为抗原-抗体的特异性作用而大量地聚集在癌细胞表面.正常的细胞由于含有微量的表面生长因子蛋白,在同样条件下进行孵育后,仅有少量的Au纳米棒结合在其表面.根据Au纳米棒聚集所具有的强烈的等离子体共振光散射信号,通过暗场显微镜观察,可以明显区分出正常细胞和癌变细胞.
Au纳米棒作为荧光探针和光散射探针在诸多领域得到了广泛的应用,克服了以传统有机荧光染料为主的荧光探针在应用中存在一些难以克服的缺陷.最近,无机发光量子点、荧光聚合物纳米微球、复合荧光二氧化硅纳米粒子等荧光纳米探针.126,135也相继出现,在一定程度上克服了传统有机荧光试剂的缺陷:有机染料等传统荧光材料的亮度不如量子点,并且存在光致褪色问题.Cho等136采用无机二氧化硅材料来阻止有机染料的光致褪色现象,为生物分析提供了新的发展领域,成为了近年来研究的热点.
3.4 表面等离子体共振技术
SPR技术原理是入射光以临界角入射到两种不同折射率的介质界面时,可引起金属自由电子的共振,由于电子吸收了光子能量,从而使反射光在一定角度内强度发生改变.137Au纳米棒等贵金属纳米颗粒所表现的与形貌、尺寸、介质环境等因素密切相关的SPR性质在上世纪80年代初首次应用于生物分子相互作用研究方面.138由于其具有可实时监测反应的动态过程,且样品无需标记和纯化的优点,现已成为生物传感、共振成像、光刻等技术领域一种强大的分析测试手段,并取得了瞩目的进展.
Van-Duyne和其合作者139,140提出基于Au纳米颗粒体系的LSPR设计生物传感器,这类生物传感器是利用LSPR引起的吸收峰位置随金属颗粒所处环境折射率的变化而发生偏移这一特性.Parab等141报导了一种利用Au纳米棒作为分子探针检测目标DNA的传感器,该传感器已成功地用于人体尿液样品中沙眼衣原体病原体基因的检测,检测浓度范围达到0.25-20 nmol·L-1.Wang等142提出基于Au纳米棒的LSPR传感器用于缓冲液、血清及血浆中乙肝病毒的含量检测.
李莹等143在传统光学方法的指纹采集系统基础上进行改进,提出了表面等离子体共振成像技术,该方法能够大幅度提高采集的指纹图像的对比度和清晰度,提高整个指纹识别系统的运行速度和识别的准确性,有望在有关个人身份认证的各个领域得到应用.
Zijlstra等3利用纳米棒的SPR波长可调性以及光学极化依赖特性,实现了高密度五维光存储,在原来三维光记录媒介的基础上增加了波长和偏振两个维度,媒介的数据密度大大提高.将纵横比不同的Au纳米棒掺入聚乙烯醇中,然后旋涂到玻璃衬底上,这样就做成了储光媒介(记录层),能在多个维度存储数据,对激光的不同波长和偏振做出响应,通过自身的双光子荧光,在材料的同一区域内多种数据图案可在互不干扰的情况下被读取和刻写.预计将在医疗、金融、军事、安全编码和银行等需要数据加密的领域获得广泛应用.144
3.5 负折射率材料
当前,关于负折射率材料的研究已经成为科学界最热门的话题之一,若材料的介电常数和磁导率同时为负值,那么材料的折射率将为负值.我们知道,在自然界发现的所有材料都具有正折射率,金属的介电常数在较宽的光波段内都是负值,但是在光波段内磁导率为负值的材料却难以找到.因此,制备负折射率材料的关键就变成能否使得人工材料的磁导率为负.
1999年,由英国Pendry等145提出了金属开口共振环结构的构想,整个材料是一种复合结构,磁共振组元使材料的有效磁导率可达到负值.2001年, Shelby等146首次在实验上证实了自然材料负折射率的存在,不过实验是在微波段(非光频段)完成的.
折射率被用来衡量电磁波从一种媒介进入另一种媒介时,光线被弯曲的程度,弯曲意味着存在光线损失.如何在更高频段内实现材料的负有效磁导率而又不带来严重的损耗,2005年,Zhang等147提出了解决这一问题的可能方案,即利用成对的金属纳米棒做为磁共振超构材料的组成单元,当光的极化方向与纳米棒的轴向平行时,会在纳米棒之间激发产生LSPR,产生电流振荡,若纳米棒对其中的电流形成反向环形电流振荡,则会在纳米棒之间的区域产生磁共振,有可能在光频范围内实现负的有效磁导率.
2010年,在研制负折射率超材料这个问题上得到突破性进展,Xiao等148将Ag和不导电的Al2O3交替层堆叠在一起,在薄膜上挖出直径100 nm的小洞,小洞交织在一起呈现出渔网图样,研制出了一种可增强光线的负折射率超材料.新研制的超材料具有改变光线传播方向的能力,光线在这种材料中会出现“负折射”,而且,因为拥有增益介质,新的光学超材料还可以增强入射的光线.将增益介质的效率提高了50倍,大大推动变换光学领域的进展.预计这种新的光学超材料有助于科学家研制出先进传感器、使用光而不是电子信号来处理信息的计算机和电子产品,甚至隐形斗篷等.
4 总结与展望
Au纳米棒独特的光学性质使其在生物医学、生化标记、成像分析、信息存储等领域有了广阔的应用前景.纳米材料在生物体内相容性的问题研究才刚刚起步,如何将神奇的纳米材料和现代技术相结合,使得Au纳米棒在生物标记、免疫检测、荧光探针、生物成像、生物传感、疾病诊断和光热治疗以及信息存储等领域应用健康发展,还需要在这个学科方向中开展更深入广泛的研究.尽管如此,古老的黄金通过现代科学的演绎,已经结出了丰硕的成果.
(1)Tian,N.;Zhou,Z.Y.;Sun,S.G.;Ding,Y.;Wang,Z.L.Science 2007,316,732.
(2) Sanvicens,N.;Marco,M.P.Trends Biotechnol.2008,26,425.
(3) Zijlstra,P.;Chon,J.W.M.;Gu,N.Nature 2009,459,410.
(4)Wang,F.;Li,C.H.;Sun,L.D.;Wu,H.S.;Ming,T.;Wang,J.F.; Yu,J.C.;Yan,C.H.J.Am.Chem.Soc.2011,133,1106.
(5) Jin,R.C.;Cao,Y.W.;Mirkin,C.A.;Kelly,K.L.;Schatz,G.C.; Zheng,J.G.Science 2001,294,1901.
(6) Millstone,J.E.;Hurst,S.J.;Métraux,G.S.;Cutler,J.I.; Mirkin,C.A.Small 2009,5,646.
(7) Romo-Herrera,J.M.;Alvarez-Puebla,R.A.;Liz-Marzán,L. M.Nanoscale 2011,3,1304.
(8) Tao,A.R.;Habas,S.;Yang,P.D.Small 2008,4,310.
(9) Kan,C.X.;Zhu,X.G.;Wang,G.H.J.Phys.Chem.B 2006, 110,4651.
(10) Li,C.C.;Sato,R.;Kanehara,M.;Zeng,H.B.;Bando,Y.; Teranishi,T.Angew.Chem.Int.Edit.2009,48,6883.
(11) Naumov,I.I.;Li,Z.Y.;Bratkovsky,A.M.Appl.Phys.Lett. 2010,96,033105.
(12) Sau,T.K.;Rogach,A.L.Adv.Mater.2010,22,1781.
(13) Xiong,Y.J.;Chen,Y.J.;Wiley,B.;Xia,Y.N.;Yin,Y.D.;Li,Z. Y.Nano Lett.2005,5,1237.
(14)Larsson,E.M.;Langhammer,C.;Zoric,I.;Kasemo,B.Science 2009,326,1091.
(15) Okamoto,H.;Imura,K.Prog.Surf.Sci.2009,84,199.
(16) Tirtha,S.;Basudeb,K.Solid State Sci.2009,11,1044.
(17) Du,S.Y.;Li,Z.Y.Opt.Lett.2010,35,3402.
(18)Yang,Z.;Ni,W.H.;Kou,X.S.;Zhang,S.Z.;Sun,Z.H.;Sun, L.D.;Wang,J.F.;Yan,C.H.J.Phys.Chem.C 2008,112, 18895.
(19) Bardhan,R.;Grady,N.K.;Cole,J.R.;Joshi,A.;Halas,N.J. ACS Nano 2009,3,744.
(20)Chowdhury,M.H.;Ray,K.;Johnson,M.L.;Gray,S.K.;Pond, J.;Lakowicz,J.R.J.Phys.Chem.C 2010,114,7448.
(21) Fang,Y.;Seong,N.H.;Dlott,D.D.Science 2008,321,388.
(22)Yoon,I.;Kang,T.;Choi,W.;Kim,J.;Yoo,Y.;Joo,S.W.;Park, Q.H.;Ihee,H.;Kim,B.J.Am.Chem.Soc.2009,131,758.
(23)Hsieh,H.Y.;Xiao,J.L.;Lee,C.H.;Huang,T.W.;Yang,C.S.; Wang,P.C.;Tseng,F.G.J.Phys.Chem.C 2011,115,16258.
(24)Shimada,T.;Imura,K.;Hossain,M.K.;Okamoto,H.M.; Kitajima,M.J.Phys.Chem.C 2008,112,4033.
(25) Nakamura,T.;Hirata,N.;Sekino,Y.;Nagaoka,S.;Nakajima,A. J.Phys.Chem.C 2010,114,16270.
(26)Wissert,M.D;Ilin,K.S;Siegel,M.;Lemmer,U.;Eisler,H.J. Nano Lett.2010,10,4161.
(27) Celebrano,M.;Biagioni,P.;Finazzi,M.;Duò,L.;Zavelani-Rossi,M.;Polli,D.;Labardi,M.;Allegrini,M.;Grand,J.; Adam,P.M.;Royer,P.;Cerullo,G.Phys.Stat.Sol.C 2008,5, 2657.
(28)Ko,K.D.;Kumar,A.;Fung,K.H.;Ambekar,R.;Liu,G.L.; Fang,N.X.;Toussaint,K.C.Nano Lett.2011,11,61.
(29) Link,S.;El-Sayed,M.A.J.Phys.Chem.B 1999,103,4212.
(30) Kelly,K.L.;Coronado,E.;Zhao,L.L.;Schatz,G.C.J.Phys. Chem.B 2003,107,668.
(31)Zoric,I.;Zach,M.;Kasemo,B.;Langhammer,C.ACS Nano 2011,5,2535.
(32) Quinten,M.Optical Properties of Nanoparticle Systems:Mie and Beyond;Wiley-VCH Verlag&Co.KgaA:Weinheim, 2011;pp 316-377.
(33) Bohern,C.F.;Huffman,D.R.Absorption and Scattering of Light by Small Particles;Wiley-VCH Verlag&Co.KgaA: Weinheim,1983.
(34)Wood,R.W.Philos.Mags.1902,4,396
(35) Mie,G.Ann.Phys.1908,25,377.
(36) Link,S.;El-Sayed,M.A.J.Phys.Chem.B 1999,103,8410.
(37) Sinzig,J.;Quinten,M.Appl.Phys.A 1994,58,157.
(38) Draine,B.T.;Flatau,P.J.J.Opt.Soc.Am.A 1994,11,1491.
(39) Brioude,A.;Jiang,X.C.;Pileni,M.P.J.Phys.Chem.B 2005, 109,13138.
(40) Kan,C.X.;Cai,W.P.;Li,C.C.;Fu,G.H.;Zhang,L.D. J.Appl.Phys.2004,96,5727.
(41) Osborn,J.A.Phys.Rev.1945,67,351.
(42) Johnson,P.B.;Christy,R.W.Phys.Rev.B 1972,6,4370.
(43)Yang,W.H.;Schatz,G.C.;Duyne,R.P.V.J.Chem.Phys. 1995,103,869.
(44) Yee,K.IEEE Trans.Antennas Propag.1966,14,302.
(45) Ghosh,S.K.;Pal,T.Chem.Rev.2007,107,4797.
(46) MurPhy,C.J.;Gole,A.M.;Stone,J.W.;Siseo,P.N.;Alkilany, A.M.;Goldsmith,E.C.;Baxter,S.C.Accounts Chem.Res. 2008,41,1721.
(47) Li,H.C.;Kan,C.X.;Yi,Z.G.;Ding,X.L.;Cao,Y.L.;Zhu,J. J.J.Nanomater.2010,doi:10.1155/2010/96271.
(48) Novo,C.;Funston,A.M.;Mulvaney,P.Nat.Nanotechnol. 2008,3,598.
(49)Manikandan,D.;Mohan,S.;Magudapathy,P.;Nair,K.G.M. Physica B 2003,325,86.
(50) Marinakos,S.M.;Chen,S.H.;Chilkoti,A.Anal.Chem.2007, 79,5278.
(51) Li,J.F.;Liu,S.Y.;Liu,Y.;Zhou,F.;Li,Z.Y.Appl.Phys.Lett. 2010,96,263103.
(52) Nie,S.;Emory,S.R.Science 1997,275,1102.
(53)LeRu,E.;Meyer,M.;Etchegoin,P.J.Phys.Chem.B 2006,110, 1944.
(54) Kim,S.;Jin,J.H.;Kim,Y.J.;Park,I.Y.;Kim,Y.;Kim,S.W. Nature 2008,453,757.
(55)Ausman,L.K.;Schatz,G.C.J.Chem.Phys.2009,131,084708.
(56) Mayer,K.M.;Hao,F.;Lee,S.;Nordlander,P.;Hafner,J.H. Nanotechnology 2010,21,255503.
(57) Gaiduk,A.;Ruijgrok,P.V.;Yorulmaz,M.;Orrit,M.Phys. Chem.Chem.Phys.2011,13,149.
(58) Kleinman,S.L.;Ringe,E.;Valley,N.;Wustholz,K.L.;Phillips, E.;Scheidt,K.A.;Schatz,G.C.;Van Duyne,R.P.J.Am.Chem. Soc.2011,133,4115.
(59)Tian,Z.Q.;Ren,B.;Wu,D.Y.J.Phys.Chem.B 2002,106, 9463.
(60) Talley,C.E.;Jackson,J.B.;Oubre,C.;Grady,N.K.;Hollars, C.W.;Lane,S.M.;Huser,T.R.;Nordlander,P.;Halas,N.J. Nano Lett.2005,5,1569.
(61) Doering,W.E.;Nie,S.J.Phys.Chem.B 2002,106,311.
(62) Lombardi,J.R.;Birke,R.L.Accounts Chem.Res.2009,42, 734.
(63) Hao,E.;Schatz,G.C.J.Chem.Phys.2004,120,357.
(64)Schatz,G.C.;Young,M.A.;Van-Duyne,R.P.Top.Appl.Phys. 2006,103,19.
(65) Kneipp,K.;Kneipp,H.;Itzkan,I.;Dasari,R.R.;Feld,M.S. Chem.Rev.1999,99,2957.
(66) Nikoobakht,B.;Wang,J.P.;El-Sayed.M.A.Chem.Phys.Lett. 2002,366,17.
(67)Nikoobakht,B.;El-Sayed,M.A.J.Phys.Chem.A 2003,107, 3372.
(68) Murphy,C.J.;Gole,A.M.;Hunyadi,S.E.;Orendorff,C.J. Inorg.Chem.2006,45,7544.
(69) Li,Z.Y.;Xia,Y.N.Nano Lett.2010,10,243
(70) Brown,L.V.;Sobhani,H.;Lassiter,J.B.;Nordlander,P.;Halas, N.J.ACS Nano 2010,4,819.
(71) Slaughter,L.S.;Wu,Y.P.;Willingham,B.A.;Nordlander,P.; Link,S.ACS Nano 2010,4,4657.
(72) Encina,E.R.;Coronado,E.A.J.Phys.Chem.C 2010,114, 16278.
(73) Feng,X.M.;Ruan,F.X.;Hong,R.J.;Ye,J.S.;Hu,J.Q.;Hu, G.Q.;Yang,Z.L.Langmuir 2011,27,2204.
(74) Barrow,S.J.;Funston,A.M.;Gomez,D.E.;Davis,T.J.; Mulvaney,P.Nano Lett.2011,11,4180.
(75) Manjavacas,A.;deAbajo,F.J.G.;Nordlander,P.Nano Lett. 2011,11,2318.
(76)Wang,Z.L.Progress in Physics 2009,29,287.[王振林.物理学进展,2009,29,287.]
(77)Koh,A.L.;Fernandez-Domínguez,A.I.;McComb,D.W.; Maier,S.A.;Yang,J.K.W.Nano Lett.2011,11,1323.
(78) Jain,P.K.;El-Sayed,M.A.Nano Lett.2007,7,2854.
(79) Jain,P.K.;El-Sayed,M.A.J.Phys.Chem.C 2008,112,4954.
(80) Encina,E.R.;Coronado,E.A.J.Phys.Chem.C 2010,114, 3918.
(81) Sheikholeslami,S.N.;Garcia-Etxarri,A.;Dionne,J.A.Nano Lett.2011,11,3927.
(82) Jain,P.K.;Eustis,S.;El-Sayed,M.A.J.Phys.Chem.B 2006, 110,18243.
(83) Funston,A.M.;Novo,C.;Davis,T.J.;Mulvaney,P.Nano Lett.2009,9,1651.
(84) Tabor,C.;Van Haute,D.;El-Sayed,M.A.ACS Nano 2009,3, 3670.
(85) Shao,L.;Woo,K.C.;Chen,H.J.;Jin,Z.;Wang,J.F.;Lin,H. Q.ACS Nano 2010,4,3053.
(86) Juluri,B.K.;Chaturvedi,N.;Hao,Q.Z.;Lu,M.Q.;Velegol, D.;Jensen,L.;Huang,T.J.ACS Nano 2011,5,5838.
(87) Alegret,J.;Rindzevicius,T.;Pakizeh,T.;Alaverdyan,Y.; Gunnarsson,L.;Kall,M.J.Phys.Chem.C 2008,112,14313.
(88) Sonnefraud,Y.;Verellen,N.;Sobhani,H.;Vandenbosch,G.A. E.;Moshchalkov,V.V.;Van-Dorpe,P.;Nordlander,P.;Maier, S.A.ACS Nano 2010,4,1664.
(89) Liu,H.;Liu,Y.M.;Li,T.;Wang,S.M.;Zhu,S.N.;Zhang,X. Phys.Status Solidi B 2009,246,1397.
(90)Chen,H.Y.;He,C.L.;Wang,C.Y.;Lin,M.H.;Mitsui,D.; Eguchi,M.;Teranishi,T.;Gwo,S.ACS Nano 2011,5,8223.
(91) Lee,S.Y.;Hung,L.;Lang,G.S.;Cornett,J.E.;Mayergoyz,I. D.;Rabin,O.ACS Nano 2010,4,5763.
(92) Lassiter,J.B.;Aizpurua,J.;Hernandez,L.I.;Brandl,D.W.; Romero,I.;Lal,S.;Hafner,J.H.;Nordlander,P.;Halas,N.J. Nano Lett.2008,8,1212.
(93) Wang,L.;Clavero,C.;Huba,Z.;Carroll,K.J.;Carpenter,E. E.;Gu,D.F.;Lukaszew,R.A.Nano Lett.2011,11,1237.
(94) Fofang,N.T.;Grady,N.K.;Fan,Z.Y.;Govorov,A.O.;Halas, N.J.Nano Lett.2011,11,1556.
(95) Lim,D.K.;Jeon,K.S.;Hwang,J.H.;Kim,H.;Kwon,S.; Suh,Y.D.;Nam,J.M.Nat.Nanotechnol.2011,6,452.
(96) Mukherjee,S.;Sobhani,H.;Lassiter,J.B.;Bardhan,R.; Nordlander,P.;Halas,N.J.Nano Lett.2010,10,2694.
(97) Xu,H.Q.;Li,H.J.;Liu,Z.M.;Xie,S.X.;Zhou,X.;Peng,X.; Xu,X.K.J.Opt.Soc.Am.A 2011,28,1662.
(98)Tabor,C.;Murali,R.;Mahmoud,M.;El-Saye,M.A.J.Phys. Chem.A 2009,113,1946.
(99)Yang,S.C.;Kobori,H.;He,C.L.;Lin,M.H.;Chen,H.Y.;Li, C.C.;Kanehara,M.;Teranishi,T.;Gwo,S.Nano Lett.2010, 10,632.
(100) Yin,P.G.;You,T.T.;Tan,E.Z.;Li,J.;Lang,X.F.;Jiang,L.; Guo,L.J.Phys.Chem.C 2011,115,18061.
(101) Fang,Z.Y.;Cai,J.Y.;Yan,Z.B.;Nordlander,P.;Halas,N.J.; Zhu,X.Nano Lett.2011,11,4475.
(102) Hao,F.;Nehl,C.L.;Hafner,J.H.;Nordlander,P.Nano Lett.2007,7,729.
(103) Aydin,K.;Pryce,I.M.;Atwater,H.A.Opt.Express 2010,18, 13407.
(104) Bao,K.;Sobhani,H.;Nordlander,P.Chin.Sci.Bull.2010,55, 2629.
(105) Marhaba,S.;Bachelier,G.;Bonnet,C.;Broyer,M.;Cottancin, E.;Grillet,N.;Lerme,J.;Vialle,J.L.;Pellarin,M.J.Phys. Chem.C 2009,113,4349.
(106) Rechberger,W.;Hohenau,A.;Leitner,A.;Krenn,J.R.; Lamprecht,B.;Aussenegg,F.R.Opt.Commun.2003,220,137.
(107) Zuloaga,J.;Nordlander,P.Nano Lett.2011,11,1280.
(108) Sheikholeslami,S.;Jun,Y.W.;Jain,P.K.Alivisatos,A.P. Nano Lett.2010,10,2655.
(109) Pena-Rodriguez,O.;Pal,U.;Campoy-Quiles,M.;Rodriguez-Fernandez,L.;Garriga,M.;Alonso,M.I.J.Phys.Chem.C 2011,115,6410.
(110) Encina,E.R.;Coronado,E.A.J.Phys.Chem.C 2011,115, 15908.
(111)Chowdhury,M.H.;Chakraborty,S.;Lakowicz,J.R.;Ray,K. J.Phys.Chem.C 2011,115,6879.
(112)Yao,H.M.;Li,Z.;Gong,Q.H.Sci.China Ser.G 2009,52, 1129.
(113) Maier,S.A.Nat.Mater.2003,2,229.
(114) Kawata,S.;Ono,A.;Verma,P.Nat.Photonics 2008,2,438.
(115) Ming,T.;Zhao,L.;Xiao,M.;Wang,J.F.Small 2010,6,2514.
(116) Imura,K.;Nagahara,T.;Okamoto,H.J.Am.Chem.Soc.2004, 126,12730.
(117) Eustis,S.;El-Sayed,M.J.Phys.Chem.B 2005,109,16350.
(118) Li,C.Z.;Male,K.B.,Hrapovic,S.;Luong,J.H.T.Chem. Commun.2005,3924.
(119)Mohamed,M.B.;Volkov,V.;Link,S.;El-Sayed,M.A.Chem. Phys.Lett.2000,317,517.
(120) Zheng,J.;Zhang,C.W.;Dickson,R.M.Phys.Rev.Lett.2004, 93,0774021.
(121) Zhu,J.;Wang,Y.C.;Yan,S.N.Chin.Phys.Lett.2004,21,559.
(122) Sun,G.M.;Yang,P.H.;Sun,J.H.;Cai,J.Y.Chin.J.Lumin. 2011,32,636.[孙桂敏,杨培慧,孙俊环,蔡继业.发光学报, 2011,32,636.]
(123) Lakowicz,J.R.;Geddes,C.D.;Gryczynski,I.;Malicka,J.; Gryczynski,Z.;Aslan,K.;Lukomska,J.;Matveeva,E.;Zhang, J.A.;Badugu,R.;Huang,J.J.Fluorescence 2004,14,425.
(124)Thomas,K.G.;Kamat,P.V.Accounts Chem.Res.2003,36, 888.
(125) Wenger,J.;Gerard,D.;Dintinger,J.;Mahboub,O.;Bonod,N.; Popov,E.;Ebbesen,T.W.;Rigneault,H.Opt.Express 2008, 16,3008.
(126) Li,X.;Kao,F.J.;Chuang,C.C.;He,S.L.Opt.Express 2010, 18,11335.
(127) Ming,T.;Zhao,L.;Yang,Z.;Chen,H.J.;Sun,L.D.;Wang,J. F.;Yan,C.H.Nano Lett.2009,9,3896.
(128) Qian,Q.;Jiang,L.;Cai,F.H.;Wang,D.;He,S.L. Biomaterials 2011,32,1601.
(129) Durr,N.J.;Larson,T.;Smith,D.K.;Korgel,B.A.;Sokolov, K.;Ben-Yakar,A.Nano Lett.2007,7,941.
(130) Wang,C.G.;Chen,J.J.;Talavage,T.;Irudayaraj,J.Angew. Chem.2009,121,2797.
(131) Wang,C.G.;Irudayaraj,J.Small 2010,6,283.
(132) Guo,H.Y.;Lu,L.H.;Wu,C.;Pan,J.G.;Hu,J.W.Acta Chim. Sin.2009,67,1603.[郭红燕,芦玲慧,吴 超,潘建高,胡家文.化学学报,2009,67,1603.]
(133) He,W.;Huang,C.Z.;Li,Y.F.;Xie,J.P.;Yang,R.G.;Zhou,P. F.;Wang,J.Anal.Chem.2008,80,8424.
(134) Huang,X.;El-Sayed,I.H;Qian,W.;El-Sayed,M.A.J.Am. Chem.Soc.2006,128,2115.
(135) Mu,X.;Wu,C.L.;Lai,J.P.;Chen,J.B.;Zheng,J.S.;Li,C.; Zhao,Y.B.Chin.Sci.Bull.2011,56,3242.
(136) Cho,E.B.;Volkov,D.O.;Sokolov,I.Adv.Funct.Mater.2011, 21,3129.
(137) Sui,S.F.;Xiao,C.D.;Yang,J.Surface Plasmon Resonance Biosensor,1st ed.;Scientific and Technical Publishers: Shanghai 2008;pp 1-44.[隋森芳,肖才德,杨 军.表面等离子体激元共振生物传感器,第一版;上海:科学技术出版社,2008:1-44.]
(138) Liedberg,B.;Nylander,C.;Lundstrom,I.Sensors and Actuators 1983,4,299.
(139) Haes,A.J.;Zou,S.L.;Schatz,G.C.;Van-Duyn,R.P.J.Phys. Chem.B 2004,108,109.
(140) McFarland,A.D.;Van-Duyne,R.P.Nano Lett.2003,3,1057.
(141) Parab,H.J.;Jung,C.;Lee,J.H.;Park,H.G.Biosens. Bioelectron.2010,26,667.
(142)Wang,X.H.;Li,Y.A.;Wang,H.F.;Fu,Q.X.;Peng,J.C.; Wang,Y.L.;Du,J.A.;Zhou,Y.;Zhan,L.S.Biosens. Bioelectron.2010,26,404.
(143) Li,Y.;Zhong,J.G.;Zhang,Y.L.Chin.J.Lasers 2006,33, 1143.[李 莹,钟金钢,张永林.中国激光,2006,33,1143.]
(144) Cao,Y.L.;Ding,X.L.;Li,H.C.;Yi,Z.G.;Wang,X.F.;Zhu, J.J.;Kan,C.X.Acta Phys.-Chim.Sin.2011,27,1273. [曹艳丽,丁孝龙,李红臣,伊兆广,王祥夫,朱杰君,阚彩侠.物理化学学报,2011,27,1273.]
(145) Pendry,J.B.;Holden,A.J.;Robbins,D.J.;Stewart,W.J. IEEE Trans.Microwave Theory Tech.1999,47,2075.
(146) Shelby,R.A.;Smith,D.R.;Schultz,S.Science 2001,292,77.
(147) Zhang,S.;Fan,W.J.;Minhas,B.K.;Frauenglass,A.;Malloy, K.J.;Brueck,S.R.J.Phys.Rev.Lett.2005,94,037402.
(148) Xiao,S.M.;Drachev,V.P.;Kildishev,A.V.;Ni,X.J.; Chettiar,U.K.;Yuan,H.K.;Shalaev,V.M.Nature 2010,466, 735.
December 9,2011;Revised:March 15,2012;Published on Web:March 16,2012.
Research Progress on the Optical Properties of Gold Nanorods
KE Shan-Lin1KAN Cai-Xia1,*MO Bo1CONG Bo1ZHU Jie-Jun2
(1Department of Applied Physics,College of Science,Nanjing University of Aeronautics and Astronautics,Nanjing 211106, P.R.China;2National Laboratory of Solid State Microstructures,Department of Physics,Nanjing University, Nanjing 210093,P.R.China)
Gold nanorods exhibit unique and tunable surface plasmon resonance(SPR)derived optical properties in the ultraviolet-visible-near infrared(UV-Vis-NIR)region.The high stability,low biological toxicity,bright color,and versatility of gold nanorods have inspired an explosion of research interest in their properties and applications(which include roles in catalysis,data storage,and biomedicine).This paper presents a brief overview of current research progress on the optical properties of gold nanorods,including surface plasmon resonance,local field enhancement,plasmon coupling,fluorescence,and application outlook.
Gold nanorod;Surface plasmon resonance;Localized field enhancement; Plasmon resonance coupling;Fluorescence
10.3866/PKU.WHXB201203162
∗Corresponding author.Email:cxkan@nuaa.edu.cn;Tel:+86-25-52113853.
The project was supported by the National Natural Science Foundation of China(51032002).
国家自然科学基金(51032002)资助项目
O647
猜你喜欢
空间科学学报(2021年6期)2021-03-09
装备制造技术(2020年4期)2020-12-25
国际呼吸杂志(2019年8期)2019-04-29
知识经济·中国直销(2018年12期)2018-12-29
当代陕西(2018年12期)2018-08-04
北京航空航天大学学报(2017年7期)2017-11-24
新农业(2017年2期)2017-11-06
光学精密工程(2016年6期)2016-11-07
中国医学装备(2015年10期)2015-12-29
太阳能(2015年12期)2015-04-12
