
肉桂酸生物法合成苯乙酮过程跟踪的定量分析方法
2014-05-14 11:01孙松马丽刘雄民关文龙
应用化工 2014年4期
孙松,马丽,刘雄民,关文龙
(广西大学化学化工学院,广西南宁 530003)
苯乙酮广泛应用于皂用香精和烟草香精等香料制造以及化妆品行业中。微生物合成苯乙酮则由于其生产过程环保,且具有良好的香气品质而备受关注[1-2]。测定肉桂酸或苯乙酮方法主要有极谱法、薄层扫描法、毛细管电泳法、高效液相色谱法、紫外分光光度法等[3-6]。其中紫外分光光度法由于其分析时间短、操作方法简便而且分析成本低廉等优点而最具应用和研究价值。但是发酵液自身的一些营养成分和菌体的紫外吸收有可能会干扰分析结果,目前主要通过额外配制加入底物前的发酵液作为参比溶液的方法扣除此类干扰[7],该方法需要额外精确配制和储备特殊参比溶液,并且对发酵液在加入底物前后的紫外吸收变化情况未做相应验证。且发酵液自身的紫外吸收由于在加入底物后难以直接测定。因此,针对其在转化过程中变化情况的相关研究也鲜有文献报道。在由肉桂酸发酵制备苯乙酮的放大生产过程中,建立一种简单快速的转化率测定方法用于转化过程的监控,具有重要的实际应用意义。
本文利用投影模拟[8]的方法进行了肉桂酸苯乙酮相对含量的检测,并将其做了延伸应用,首次利用该方法验证了30 L发酵罐中菌株Mucor sp.发酵液自身紫外吸收在加入底物后的转化过程中无明显变化;并通过测定转化初期肉桂酸含量的测量值与真实值(100%)之间的校正系数来扣除发酵液自身的干扰,将成分复杂的转化液检测简化为简单的肉桂酸和苯乙酮二组分检测。该方法简单有效,无需额外配制和储备特殊参比溶液。且该方法的验证和测量用转化液样品取自30 L中试用发酵罐,比传统实验中的摇瓶发酵更接近工业生产条件,因此,十分适用于微生物发酵工业生产中的条件优化以及转化率的快速检测和过程跟踪。另外,本文还将该方法与液相色谱法进行了比较。
1 实验部分
1.1 试剂与仪器
苯乙酮、肉桂酸、无水乙醇均为分析纯;实验菌种为本课题组筛选得到的菌种。
UV-2550型紫外-可见分光光度计;SPD-10A UV-VIS高效液相色谱仪(LC-20AT泵);色谱柱Alltima C18(250 mm ×4.6 mm,5 μm)。
1.2 溶液配制
1.2.1 肉桂酸和苯乙酮标准品溶液的制备 精确称取相同摩尔浓度的肉桂酸和苯乙酮标准品(分别为0.261 8 g和0.212 2 g),以无水乙醇溶解定容到100 mL的棕色容量瓶,分别配制成等摩尔浓度溶液Ⅰ和Ⅱ,低温保存备用。
1.2.2 转化液的制备 将菌株 Mucor sp.接种于30 L发酵罐中,在适合的培养条件下培养48 h,得到发酵液。再加入90 g左右肉桂酸进行转化,得到转化液。
1.3 转化液的液相色谱分析
准确移取转化液2 mL,用无水乙醇稀释定容至10 mL。离心,取上清液进行高效液相色谱分析。色谱条件:流动相为乙腈 ∶水 ∶冰醋酸=65∶35∶0.05;检测波长 249 nm;流速 1.0 mL/min。
2 结果与讨论
2.1 肉桂酸、苯乙酮及其混合物的紫外光谱特性
溶液Ⅰ和Ⅱ与肉桂酸相对摩尔含量0,20%,40%,60%,80%,100%的比例依次混合,得到6份总摩尔浓度相同的肉桂酸和苯乙酮混合溶液各10 mL。取20 μL再稀释至10 mL后进行紫外扫描,结果见图1。曲线1和曲线6分别为100%肉桂酸和100%苯乙酮的紫外吸收。
由图1可知,肉桂酸和苯乙酮的最大吸收波长分别为272 nm和241 nm,曲线1~6交于O点,该点为两化合物的等吸收点,波长为249 nm,当总摩尔浓度为3.53 ×10-5mol/L 时,吸光度为0.317 9。以标准溶液1~6在272 nm(此处吸光度差值最大,故选择272 nm为检测波长)的吸光度A272对肉桂酸相对摩尔含量X进行线性回归,可得肉桂酸相对摩尔含量的回归方程:

由于式(1)是在标准溶液等吸收点处的吸光度为0.317 9时得出,而待测样品的吸收曲线①不满足该方程的使用条件,因此需将样品稀释,得吸收曲线②,再利用等比例关系,模拟[8]出样品在等吸收点处吸光度为0.317 9的吸收曲线③,吸收曲线③符合式(1)使用条件,此时利用式(1)即可求出肉桂酸相对摩尔含量。另由于发酵液自身存在紫外吸收,故式(1)不直接适用于转化液中肉桂酸相对摩尔含量的测定,需扣除发酵液自身吸收干扰。
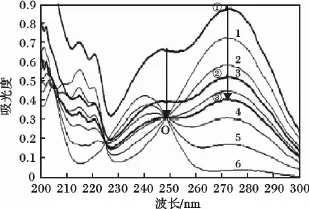
图1 总摩尔浓度相同但相对摩尔含量不同的肉桂酸和苯乙酮混合物的紫外光谱Fig.1 UV spectrum of mixture of CMA and APO with the same molar fraction but different total molar concentration
2.2 转化液定量分析方程的建立
2.2.1 发酵液自身紫外吸收变化验证 于30 L发酵罐中分别移取适量转化前(6 h)、中(72 h)、后期(96 h)转化液,稀释到适合浓度后测量其紫外吸收曲线,利用2.1节的方法模拟至同等吸收点后得到转化前、中、后期吸收曲线1,2,3。再利用等比例关系,将曲线1,2投影模拟出曲线3',见图2。
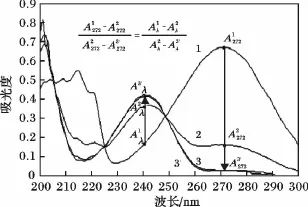
图2 由转化前期曲线1与中期曲线2通过比例关系投影出的曲线3'与后期实际吸收曲线3Fig.2 UV spectrum of fermentation broth in initial,middle and late stage(spectrum 1,2,3)and theoretical spectrum 3'
由图2可知,模拟出的吸收曲线3与实际吸收曲线3相吻合,表明发酵液的自身吸收在转化的过程中无明显变化,否则会由于受到发酵液自身吸收变化的影响,由前、中期投影模拟出的后期吸收曲线会与实际后期吸收曲线存在较大偏移。因此,该菌株发酵液自身紫外吸收在整个转化过程中变化可忽略。
2.2.2 发酵液自身吸收干扰的扣除及总分析方程的建立 由于菌株Mucor sp.发酵液在转化过程中其发酵液的背景吸收曲线无明显变化,则可认为其在波长为272 nm处吸光度A0保持不变。式(1)可以变为:

其中,A0/0.697 5在整个转化过程中不变,即为校正系数,在转化初期(<6 h)酶处于诱导时期时取样,此时肉桂酸未转化,含量为100%,校正系数可用下式求出:

联立式(2)、(3),得出转化液定量分析方程:
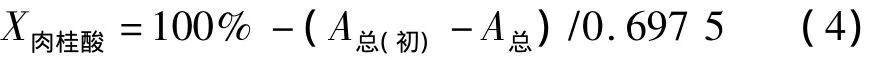
其中,A总与 A总(初期)均为转化液经稀释模拟回归至等吸收点为0.317 9时272 nm处的吸光度。

2.3 生物转化液中肉桂酸和苯乙酮相对摩尔含量(转化率)的测定
于转化初期取样,用无水乙醇稀释至合适浓度后测量其249 nm与272 nm处吸光度A272(样品)(初期)与 A249(样品)(初期),再通过稀释,测得其稀释液吸光度 A272(稀释)(初期)和 A249(稀释)(初期),通过式(5)计算出 A总(初期)。
相同方法取样,测试计算任意转化时间吸光度A总,最后利用式(4)即可得出该转化时刻肉桂酸相对摩尔含量X肉桂酸,苯乙酮相对摩尔含量(即转化率)为100% -X肉桂酸。
2.4 精密度实验
配制适宜质量浓度的肉桂酸和苯乙酮的标准品混合溶液25 mL,平行测定6次,结果见表1。
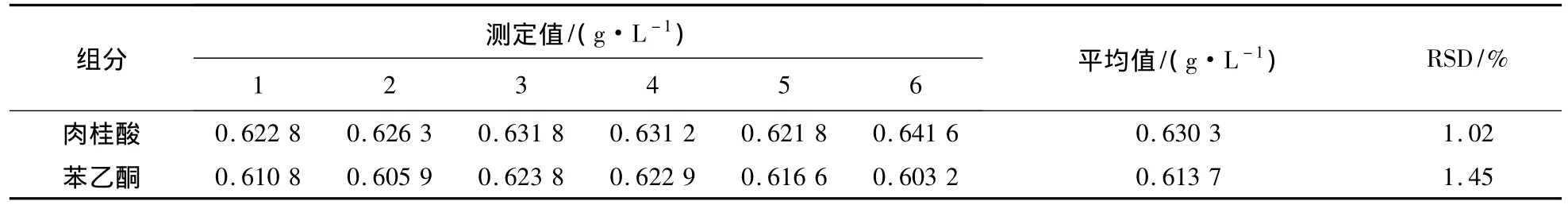
表1 方法的精密度Table 1 Precision of analysis method
由表1可知,两者的重复性都较高,标准偏差(RSD)分别为 1.02%和 1.45%。
2.5 稳定性实验
配制适宜质量浓度的肉桂酸和苯乙酮的标准品混合溶液25 mL,室温下放置,在24 h内测定溶液肉桂酸和苯乙酮的含量,结果见表2。
由表2可知,在24 h内两者均比较稳定,标准偏差(RSD)分别为4.94%和4.07%。
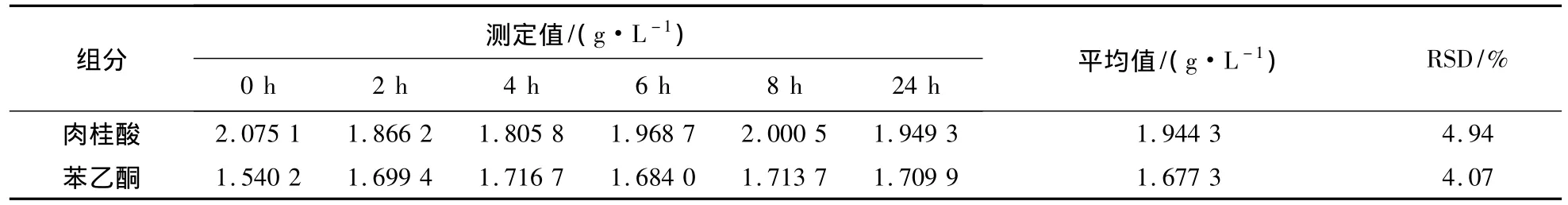
表2 方法的稳定性Table 2 Stability of analysis method
2.6 回收率实验
取3份相同已知浓度的样品,分别添加低、中、高浓度的肉桂酸和苯乙酮标准品溶液,分别测定加标样品中肉桂酸和苯乙酮的浓度(等吸收点测总浓度,式(1)测含量),结果见表3。
由表3可知,肉桂酸和苯乙酮的平均回收率分别为101.34%和99.94%,标准偏差分别为1.53%和 0.46%。
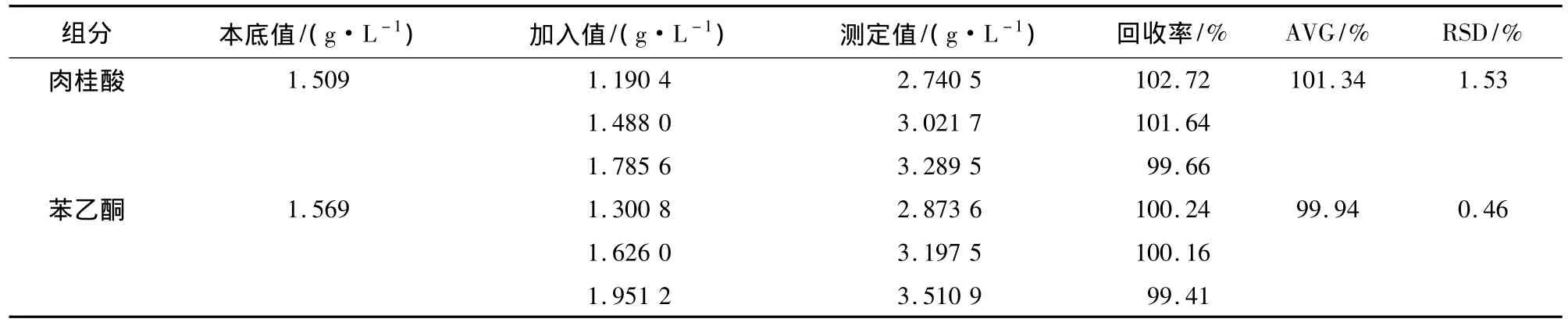
表3 方法的回收率Table 3 Recovery of analysis method
2.7 生物转化样品的测定
将1份转化液样品稀释到合适浓度后,使用上述UV法和HPLC分别测定3次,结果见表4。
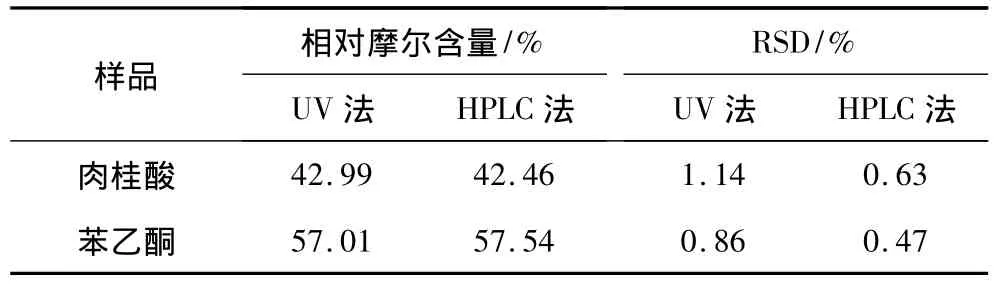
表4 样品的测定与t值检验Table 4 Determination of sample and t value test
经统计学的t检验,所得t值为1.92,小于结果的临界值2.92(α=0.05),表明这两种方法无显著性差异,说明所建立的分析方法准确可靠。
3 结论
针对微生物转化液的紫外检测时不可避免的发酵液自身的紫外吸收干扰问题,开展了生物转化过程的复杂体系定量分析方法研究,得到了如下结论:
(1)投影模拟法证明发酵液在转化期间其自身吸收无明显变化后,通过初期取样可获得校正系数,可通过该校正系数扣除发酵液自身干扰。
(2)肉桂酸相对含量与吸光度关系为:X肉桂酸=100% - (A总(初)- A总)/0.697 5,苯乙酮相对含量(即转化率)与吸光度关系为:X苯乙酮=(A总(初)-A总)/0.697 5。A总与 A总(初期)均为转化液经稀释模拟回归至等吸收点为0.317 9时272 nm处的吸光度。
(3)用t检验法将新建UV法与HPLC法进行判断,两种方法无显著性差异。
(4)该方法的验证与检测所用转化液样品取自30 L中试用发酵罐,更接近工业生产条件。因此,该方法十分适用于微生物发酵工业生产中的条件优化以及转化率的快速检测和过程跟踪。
[1]谢建春,何坚.苯乙酮及其缩酮香料化合物合成的演变[J].北京日化,1993(2):5-8.
[2]马丽,刘雄民,庞宗文.Mucor sp.JX23发酵液生物催化肉桂酸降解生成苯乙酮[J].食品与发酵工业,2010,36(1):8-10.
[3]Schrader J,Etschmann M M W,Sell D,et al.Applied biocatalysis for the synthesis of natural flavour compounds current industrial process and future prospects[J].Biotech Letters,2004,26:463-472.
[4]刘岳树,马武生.气相色谱法同时测定过氧化氢异丙苯中异丙苯和苯乙酮[J].分析科学学报,2010(6):114-118.
[5]周培文,夏炽中,冯育林.薄层色谱扫描法测定四氢叶酸辅酶模型与格氏试剂反应的产物——苯乙酮和二苯甲酮[J].山西大学学报:自然科学版,1990(2):356-361.
[6]王建刚,黄文焕,李健秀,等.高效液相色谱法测定从酚焦油裂解液中回收的苯乙酮含量[J].化学工程师,2004(7):27-28.
[7]马丽,韦一萍,刘雄民.双波长紫外分光光度法测定生物转化液中苯乙酮和肉桂酸的含量[J].食品工业科技,2009,30(2):336-337.
[8]卢志泉,刘雄民,朱丽芳,等.反式茴脑生物合成茴香酸体系紫外分光光度法的测定方法研究[J].应用化工,2013,42(4):734-738.
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
食品安全导刊(2022年11期)2022-05-17
世界科学技术-中医药现代化(2021年10期)2021-03-02
理化检验-化学分册(2020年2期)2020-06-01
西南石油大学学报(自然科学版)(2019年5期)2019-12-20
消费导刊(2019年14期)2019-08-21
消费导刊(2019年27期)2019-07-22
天然产物研究与开发(2018年4期)2018-05-07
中成药(2018年1期)2018-02-02
中国油脂(2017年6期)2017-07-25
