
密度泛函理论计算GanZn(n=1~7)团簇的几何结构和稳定性
2014-07-13 03:39贺德春
原子与分子物理学报 2014年3期
贺德春
(河西学院物理与机电工程学院,张掖734000)
1 引 言
团簇是指几个甚至成千上万个原子、分子或离子通过一定的物理或化学结合力组成的相对稳定的微观或亚微观聚集体[1].其性质不同于单个原子和分子,也不同于固体或液体.它是各种物质由原子和分子向体相物质转变的中间过渡态,它的很多新的物理和化学性质日益引起人们的关注[2-6].近年来,过渡金属团簇由于在电子器件、储氢材料等方面潜在的应用价值,而被科学家在理论和实验上进行了大量研究[7-9],而将一个金属原子掺杂到一个纯团簇中,会改变纯团簇的几何结构、稳定性和电子特性[10,11].
镓团簇由于其自身奇特的物理性质在微电子和光电子领域有着广泛应用,使人们在理论和实验上对其物理和化学性质进行了大量的研究[12-19].BelBruno[17]应用密度泛函理论计算了Gan(n=1-5)团簇的电子性质、几何结构和结合能.Song 和Cao[18]同样应用密度泛函理论研究了Gan(n=2-26)团簇的几何结构和电子性质,研究表明,随着团簇原子序数的增加,镓团簇越来越趋于紧凑结构.团簇单原子结合能随团簇尺寸的增大而增大,但是Ga8、Ga14和Ga20表现出更高的稳定性.所有偶数的镓团簇是闭壳单重态,但Ga2和Ga4团簇基态出现自旋极化现象.对于Ga2,Albe[12]计算得到它的半径r0=2.3235,结合能D0=1.4eV,频率ω0=162cm-1,而在实验上,Huber K.和Herzberg G.
[19]得到Ga2的结合能D0=1.4eV,频率ω0=162 cm-1,计算结果和实验值几乎一致.在实验上,对镓团簇其他性质的研究也比较多,比如Cha ChiaYen[20]等研究了Ga2团簇的光电子质谱,Tan和Dagdigian[13]测量了Ga2团簇的电子谱,Froben[14]研究了Ga2团簇的拉曼谱.
上面列举了对镓纯团簇的研究,随着镓团簇在材料学中的广泛应用,对掺杂镓团簇的研究也进行了大量研究[21-24].Neal[21]研究了Al掺杂到Ga+n(n=17,19,20,30-33,43,46,47)团簇的热量测定,研究表明代替一个铝原子对整个团簇的熔化性能影响很小.李恩玲等利用密度泛函理论对GanP和GanP2(n=1~7)团簇的几何结构、电子态和稳定性进行了研究,研究表明,当n≤5时,团簇的几何结构基本上为平面结构,当n>5时,团簇均为立体结构.可以看出,当对镓团簇掺杂不同的原子时,得到的新团簇的几何结构、电子态及稳定性与纯镓团簇相比,都发生了很大的变化.锌由于具有优良的物理特性,而被广泛用在发光二极管、激光器、压电材料、化学传感器和太阳能电池等方面.Tanaka 等[25]研究了Au5Zn+团簇的芳香性.Koyasu等[26]研究了AunZn-(n=2-7)团簇的光电子谱,研究表明,由于金团簇和掺杂的锌原子之间的相互作用,团簇的电子亲和势和特征光谱随着n的变化而呈现一种奇偶交替的现象.在本文,我们用DFT 中的B3LYP 方法,在LANL2DZ基组水平上,对GanZn(n=1~7)团簇的各种可能结构进行了优化,得到团簇的一系列稳定结构,分析团簇的生长规律、稳定性和磁性随团簇尺寸的变化规律.
2 计算方法
采用Gaussian 03程序密度泛函进行计算,用含有电子相关效应校正的DFT 中的B3LYP 方法,该方法是由Becke 建议的杂化函数和Lee-Yang-Parr相关函数组成,选择LANL2DZ 基组.为了确定所选方法的可靠性,我们首先计算了Ga2,Zn2和GaZn团簇的键长,频率和结合能,并与实验结果和其他理论计算进行了比较,其比较结果列于表1中.本文计算得到的Ga2团簇的键长为2.8626Å,频率为143.1299cm-1,离解能为1.165 eV.从表中可以看到,与其他理论计算得到的Ga2团 簇 的 键 长 Re=2.746 Å[27],频 率ωe=162cm-1[27],离解能De=1.13eV[28]的结果非常接近,并且从表中可以看到,本文计算Ga2团簇的结果与实验测量得到的结果也非常接近(实验结果为ωe=165cm-1,De=1.4eV)[19].对于Zn2团簇,本文计算得到的键长为4.3602Å,频率为184.9711 cm-1,离解能为0.0027eV,对GaZn团簇,本文计算得到的键长为3.0434Å,频率为75.9816cm-1,离解能为0.1412eV.对于Zn2团簇和GaZn团簇,本文没有查找到其他文献的计算结果和实验结果.可见,本文选择的方法比较适合计算Zn掺杂到Ga团簇中.
在优化GanZn(n=1~7)团簇的过程中,我们参考了许多已研究过的过渡金属团簇的结构,特别是研究了纯的Gan团簇,并且每一种结构我们考虑了不同的自旋多重度.使用同样的方法和基组,对每一种结构作了频率计算,以确保所给出的结构是稳定的,所计算的结构中不含虚频.
3 结果和讨论
3.1 几何结构
采 用DFT 中 的B3LYP/LANL2DZ 方 法 对GanZn(n=1~7)团簇所有可能的几何结构进行键长和键角的全面优化,根据能量最低原理得到了GanZn(n=1~7)团簇的基态稳定结构,其结构图如图1所示.图中n 表示Ga原子的个数,a,b,c……等表示原子数相同时,不同的结构.对所有的这些结构,它们的点群、自旋多重度、键长(Ga原子和Ga原子之间的最小键长和Ga原子与Zn原子之间的最小键长)、频率和平均键能列于表2中.
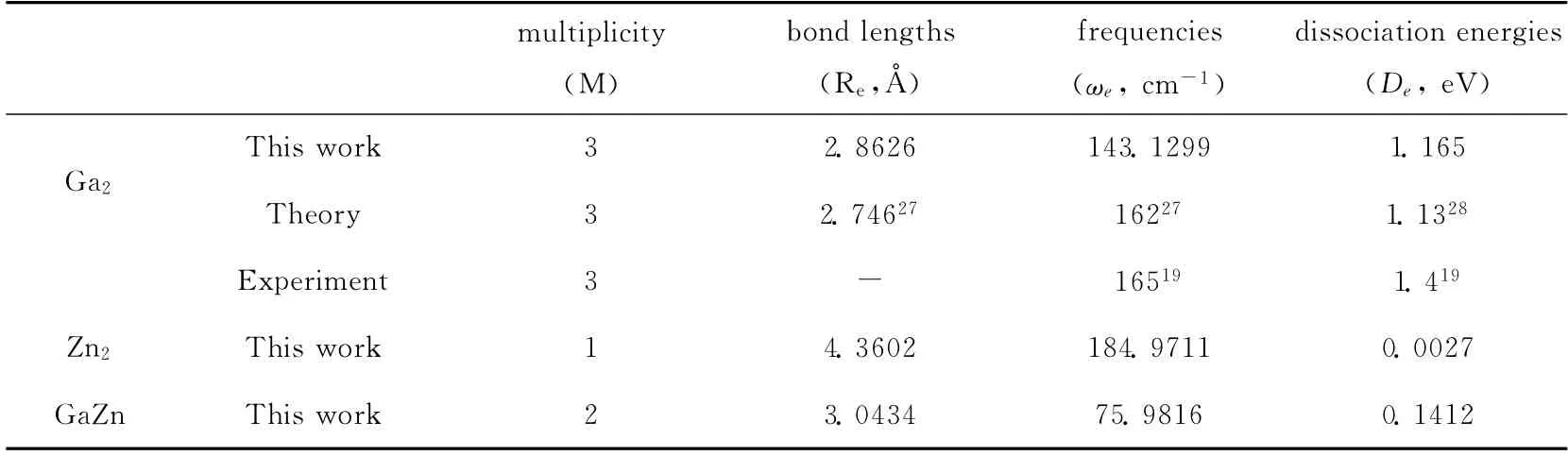
表1 团簇Ga2,Zn2 和GaZn的多重度(M),键长(Re,Å),频率(ωe,cm-1)和离解能(Dissociation Energies)(包括本工作,实验值和其他理论计算)Table 1 The computed bond lengths(Re),vibrational frequencies(ωe),and dissociation energies(De)of dimers(Ga2,Zn2and GaZn)and available experimental and previous theoretical data

图1 稳定的GanZn(n=1~7)团簇的几何结构Fig.1 The stable geometries of GanZn(n=1~7)clusters
从图1中可以看到,对于Ga1Zn团簇,它是线性分子,属于Cv群,优化平衡结构的键长为3.043Å,振动频率为75.982cm-1,它的离解能为0.071eV.Ga2Zn团簇的最稳定结构是三角形构型(C2v),Ga-Zn之间的键长为2.769Å,Ga-Ga之间的键长为2.960Å,离解能为0.508eV.同时Ga2Zn团簇还有两个线性结构,分别为Zn原子在两个Ga原子一侧和在两个Ga原子之间,这两个线性结构的稳定性都比三角结构的稳定性差.Ga3Zn团簇的最稳定的构型不是正四面体,而是发生了形变的Cs的四面体,它的自旋多重度为2,离解能为0.672eV,Ga2Zn团簇中其铲形结构的稳定性最差,而另一个四面体结构(3-a)的稳定性居中.Ga4Zn团簇的各种构型,我们共计算得到了6种构型,其中4-f是最稳定的结构,其能量比4-a,4-b,4-c,4-d和4-e分别高出0.782eV,1.324eV,2.884eV,0.002eV 和0.104eV,它的自旋多重度为1,最小的Ga-Zn键长和Ga-Ga键长相等,是2.745Å,其离解能为0.931eV.优化得到的Ga5Zn团簇的构型有三种,最稳定的构型是具有C1对称性的面心戴帽四方锥结构(5-b),它的自旋多重度为2,离解能为1.046eV,Ga-Zn键长为2.671Å,Ga-Ga键长为2.674Å.另两个亚稳态结构为自旋六重态的5-a和自旋多重态的5-c,它们的离解能分别为0.911eV 和0.826 eV.通过对Ga6Zn团簇的各种结构的优化,我们总共得到了8种Ga6Zn团簇,其中最稳定的是6-h,它的自旋多重态是1,属于C2群,其他七种结构均为亚稳态结构.6-h的离解能为1.277eV,6-a,6-b,6-c,6-d,6-e,6-f和6-g的离解能分别为0.849 eV,1.230 eV,1.084 eV,0.948 eV,0.986eV,1.101eV 和1.261eV.对 于Ga7Zn 团簇,我们得到的最稳定结构为7-a,它具有C1对称性的一个自旋二重态,最小的Ga-Zn 键长为2.726Å,最小的Ga-Ga键长为2.573Å,离解能为1.240eV.另外得到了三中亚稳态结构,分别为7-b,7-c 和7-d,其 能 量 比7-a 分 别 高 了0.237eV,0.474eV 和0.373eV.
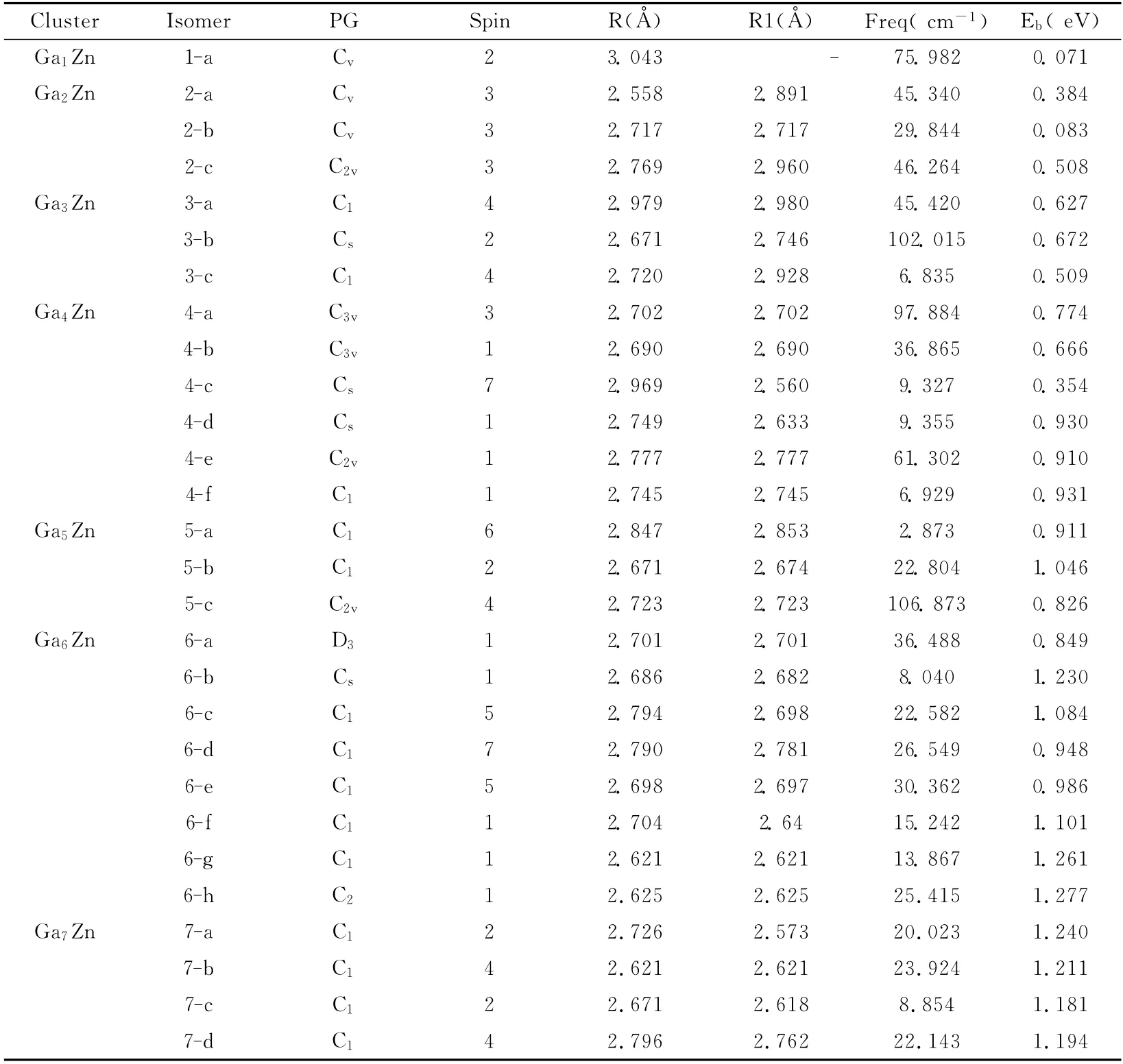
表2 团簇GanZn(1-7)的点群、自旋多重度、键长(Ga原子和Zn原子之间的最小键长和Ga原子与Ga原子之间的最小键长)、频率(最低的振动频率)和平均键能Table 2 The point group symmetries(PG),spin multiplicity,bond lengths,vibrational frequencies,and average binding energies of GanZn(1-7)clusters.R and R1denote the shortest Ga-Zn and Ga-Ga bond length,respectively;Freq denotes the lowest vibrational frequency of GanZn equilibrium geometry
3.2 GanZn(n=1~7)团簇的稳定性分析
在团簇的研究中,最基本也是最重要的是找出团簇的基态结构和相对稳定性,前面,我们确定了GanZn(n=1~7)团簇的基态结构,为了分析团簇的稳定性随团簇尺寸的关系,我们分析了GanZn(n=1~7)团簇的平均键能(Eb),能量的二阶差分能(Δ2E)和分裂能(Ef).平均键能、二阶差分能和分裂能的计算公式分别是:
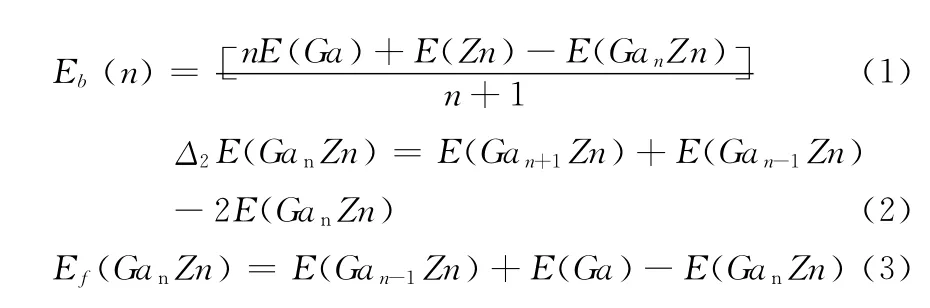
以上三个式中,E(Ga)和E(Zn)分别代表自由Ga原 子 和 自 由Zn 原 子 的 能 量,E(Gan-1Zn)、E(GanZn)和E(Gan+1Zn)分别代表最稳定团簇的总能.通过计算团簇的平均键能可以了解团簇间的相对稳定性,图2给出了团簇GanZn(n=1~7)的平均键能随团簇原子个数的变化曲线.从图2中可以看出,团簇GanZn(n=1~7)的平均键能随原子个数的增加而单调增大,这表明团簇在生长过程中不断得到能量,但增幅随着原子个数的增大在逐渐减小,原因是随着团簇原子个数的增大平均键长增加,稳定性会降低,为了维持整个团簇的稳定性,使每个原子尽可能多的与其他原子成键,从而导致了团簇配位数的增加,团簇的稳定是在键长和配位数共同作用下达到平衡的结果.特别地,原子数从n=1变化到n=6的过程中,平均键能的增长率是较大的,从n=6到n=7曲线变得平缓,在n=2和n=6 时,曲线有个小突起.从平均键能的变化曲线,我们可以粗略地说,团簇Ga2Zn和团簇Ga6Zn相对于各自相邻的团簇更稳定.

图2 GanZn(n=1~7)团簇的平均键能Eb 随n的变化Fig.2 Binding energy Ebof GanZn(n=1~7)clusters with n=1~7
接着我们又分析了团簇GanZn(n=1~7)的二阶差分能,团簇的二阶差分能是衡量基态团簇的相对稳定最直观有效的分析方法,二阶差分能的值越大,相应的结构就越稳定.图3显示了团簇GanZn(n=1~7)的二阶差分能随着原子个数的增加的变化曲线,从图中可以看出,二阶差分能随着Ga原子数的变化,出现奇偶交替的现象,当n=1,3,5等奇数时,二阶差分能为谷值,当n=2,4,6等偶数时,二阶差分能为峰值,表明n为偶数比奇数结构更为稳定,在所有计算的团簇中,Ga6Zn团簇的二阶差分值最大,表明它的稳定性最高.
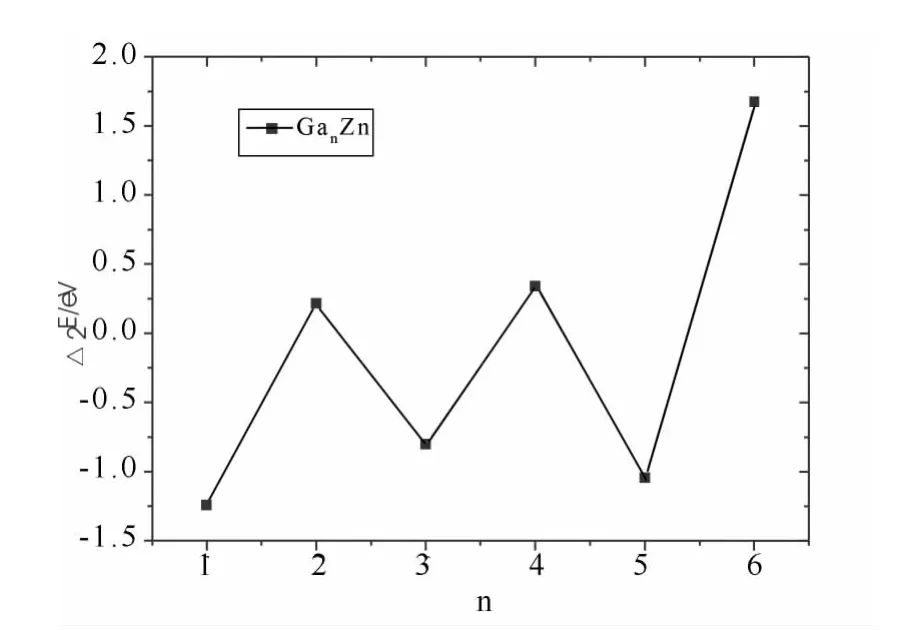
图3 GanZn(n=1-6)团簇的二阶差分能Δ2E随n的变化Fig.3 Second-order difference of total energiesΔ2Eof GanZn(n=1~7)clusters with n=1-6
为了更进一步确定团簇GanZn(n=1~7)的稳定性,我们计算了分裂能(Ef),图4给出团簇GanZn(n=1~7)的分裂能随原子个数的变化情况.分裂能表示团簇的热学稳定性,分裂能越大表示该团簇的热学性质越稳定.从图中可以看出,团簇Ga2Zn,Ga4Zn和Ga6Zn具有更高的分裂能,再一次说明它们具有更高的稳定性,团簇GaZn,Ga3Zn,Ga5Zn和Ga7Zn具有较低的分裂能,再一次说明它们具有较差的稳定性.这个结论与前面得到的二阶差分能的变化趋势相同,即当n=1,3,5,7等奇数时,团簇的稳定性差,而n=2,4,6等偶数时,团簇的稳定较好,同样表现出奇偶交替的现象.
4 结 论
采用密度泛函理论中的B3LYP/LANL2DZ泛函研究了团簇GanZn(n=1~7)的结构和稳定性.对团簇所有可能的几何结构进行了键长和键角的全面优化,对每一种结构都考虑了自旋多重态,最终得到每一种团簇的最稳定结构.通过计算团簇GanZn(n=1~7)的平均键能(Eb)、二阶差分能(Δ2E)和分裂能(Ef)来衡量稳定性.计算结构表明:
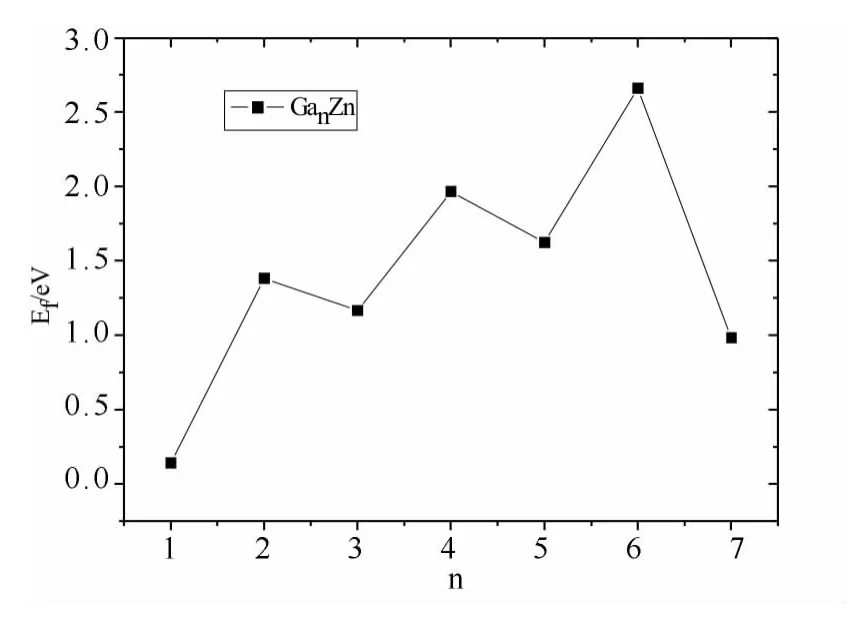
图4 GanZn(n=1~7)团簇的分裂能(Ef)随n的变化Fig.4 Fragmentation energies(Ef)of GanZn(n=1~7)clusters with n=1~7
(1)优化了大量的GanZn(n=1~7)团簇的初始结构,得到了每一种团簇的最稳定结构.
(2)研究了GanZn(n=1~7)团簇中最稳定团簇的平均键能(Eb)、二阶差分能(Δ2E)和分裂能(Ef),计算结果表明,团簇GanZn(n=1~7)的平均键能随原子个数的增加而单调增大;当n=1,3,5等奇数时,二阶差分能为谷值,当n=2,4,6等偶数时,二阶差分能为峰值;团簇Ga2Zn,Ga4Zn 和Ga6Zn具有更高的分裂能,说明它们具有更高的稳定性,团簇Ga1Zn,Ga3Zn,Ga5Zn 和Ga7Zn 具有较低的分裂能,说明它们具有较差的稳定性.
[1] Wang G H.Recent progress on cluster physics(I)[J].Progress Phys.,1994,14(2):121(in Chinese)[王广厚.团簇物理的新进展(I)[J].物理学进展,1994,14(2):121]
[2] Du J G,Sun X Y,Jiang G.Structures,chemical bongding,magnetisms of small Al-doped zirconium clusters[J].Phys.Lett.A,2010,374:854.
[3] Hua Y W,Liu Y L,Jiang G,et al..Geometric transition and electronic properties of titanium-doped aluminum clusters:AlnTi(n=2-24)[J].J.Phys.Chem.,2013,117:2590.
[4] Hu C H,Chizallet C,Toulhoat H,et al.Structural,energetic,and electronic trends in low-dimensional late-transition-metal systems[J].Phys.Rev.B,2009,79:195416.
[5] Gao A M,Li G L,Chang Y,et al.Theoretical studies on the structures and properties of As-doped Sin(n=1-8)clusters[J].J.Mole.Struct.:Theochem,2010,961:88.
[6] Zhao W J,Yang Z,Yan Y L,et al.Ground state structures and magnetisms of GenFe (n=1-8)clusters:The density functional investigations[J].Acta Phys.Sinica,2007,56(5):2596 (in Chinese)[赵文杰,杨致,闫玉丽,等.密度泛函理论计算GenFe(n=1-8)团簇的基态结构及其磁性[J].物理学报,2007,56(5):2596]
[7] Wang S,Liu Z P,Lu J,et al.Study on the size effects of Cun(n≤20)clusters with combined density functional and genetic algorithm methods[J].Acta Chem.Sinica,2007,65(17):1831(in Chinese)[王顺,刘智攀,陆靖,等.用密度泛函和遗传算法研究Cun(n≤20)团簇的尺寸效应[J].化学学报,2007,65(17):1831]
[8] Wang J,Han J G.A theoretical study on growth patterns of Ni-doped germanium clusters[J].J.Phys.Chem.B,2006,110:7820.
[9] Tian F Y,Jing Q,Wang Y X.Structure,stability,and mangnetism of ScnAl(n=1-8,12)clusres:Density functional theory investigations[J].Phys.Rev.A,2008,77:013202.
[10] Li Y F,Kuang X Y,Wang S J,et al.Geometries,Stabilities,and electronic properties of small Mgdoped gold clusters:A density functional dheory study[J].J.Phys.Chem.A,2010,114:11691.
[11] Yang Z,Yan Y L,Zhao W J,et al.Structures and magnetism of FeBn(N≤6)clusters[J].Acta Phys.Sinica,2007,56(5):2590(in Chinese)[杨致,闫玉丽,赵文杰,等.FeBn(N≤6)团簇的结构与磁性[J].物理学报,2007,56(5):2590]
[12] Albe K,Nordlund K,Nord J,et al.Modeling of compound semiconductors:Analytical bond-order potential for Ga,As,and GaAs[J].Phys.Rev.B,2002,66:035205.
[13] Tan X F,Dagdigian P J.Electronic spectrum of the gallium dimmer[J].J.Phys.Chem.A,2003,107:2642.
[14] Froben F W,Schulze W,Kloss U.Raman spectra of matrix-isolated group IIIA dimers:Ga2,In2,Ti2[J].Chem.Phys.Lett.,1983,99(5):500.
[15] Breaux G A,Hillman D A,Neal C M,et al.Gallium cluster“magic melters”[J].J.Am.Chem.Soc.,2004,126:8628.
[16] Breaux G A,Benirschke R C,Sugai T,et al.Hot and solid gallium clusters:too small to melt[J].Phys.Rev.Lett.,2003,91(21):215508.
[17] BelBruno J J.Bonding and energetics in small clus-ters of gallium and arsenic[J].Heteroatom Chem.,2003,14(2):189.
[18] Song B,Cao P L.Evolution of the geometrical and electronic structures of Gan(n=2-26)clusters:A density functional theory study[J].J.Chem.Phys.,2005,123:144312.
[19] Huber K,Herzberg G.Constants of Ddiatomic molecule[M].Van Nostrand Reinhold,New York,1979.
[20] Cha C Y,Ganteför G,Eberhardt W.The development of the 3p and 4p valence band of small aluminum and gallium clusters[J].J.Chem.Phys.,1994,100(2):995.
[21] Neal C M,Starace A K,Jarrold M F.Melting of alloy clusters:Effects of aluminum doping on gallium cluster melting[J].J.Phys.Chem.A,2007,111:8056.
[22] Li E L,Yang C J,Chen G C,et al.First principles study on structure and stability of small GanPmclusters[J].Acta Phys.Sinica,2005,54(9):4117(in Chinese)[李恩玲,杨成军,陈贵灿,等.第一性原理对GanPm小团簇的结构及稳定性的研究[J].物理学报,2005,54(9):4117]
[23] Zhang W Q,Hong X H,Zhao G F.Geometrical and electronic structures of GanP(n=1~7)clusters[J].J.Changshu Institute Tech.(Natural Sci-ences),2009,23(8):42(in Chinese)[张文庆,洪新华,赵高峰.GanP(n=1~7)团簇的几何结构和电子性质[J].常熟理工学院学报(自然科学),2009,23(8):42]
[24] Zhang F Y,Liu F L.Density functional theory study on the structure and property of CunGa(n=1-3)clusters[J].Natural Sciences J.Harbin Normal Univ.,2008,24(1):35(in Chinese)[张凤云,刘凤丽.CunGa(n=1-3)二元合金小团簇结构和稳定性的密度泛函研究[J].哈尔滨师范大学自然科学学报,2008,24(1):35]
[25] Tanaka H,Neukermans S,Janssens E,et al.σaromaticity of the bimetallic Au5Zn+cluster[J].J.Am.Chem.Soc.,2003,125:2862.
[26] Koyasu K,Naono Y,Akutsu M,et al.Photoelectron spectroscopy of binary Au cluster anions with a doped metal atom:AunM-(n=2-7),M=Pd,Ni,Zn,and Mg[J].Chem.Phys.Lett.,2006,422:62.
[27] Das K K.Ab initio MRD-CI study of the electronic states of the gallium dimmer[J].J.Phys.B:At.Mol.Opt.Phys.,1997,30:803.
[28] Roos B O,Lindh R,Malmqcist PÅ,et al.Main group atoms and dimers studied with a new relativistic ANO basis set[J].J.Phys.Chem.A,2004,108:2851.
猜你喜欢
小学生学习指导(低年级)(2021年9期)2021-10-14
应用数学(2020年2期)2020-06-24
数学物理学报(2020年1期)2020-04-21
中学生数理化·七年级数学人教版(2019年10期)2019-11-25
小学生学习指导(低年级)(2019年9期)2019-09-25
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中央民族大学学报(自然科学版)(2018年3期)2018-11-09
小学生学习指导(低年级)(2018年9期)2018-09-26
枣庄学院学报(2015年5期)2016-01-09
