
3-卤代氧化吲哚的合成及其在构建3,3-二取代氧化吲哚反应中的应用*
2014-08-15 10:49赵建强张晓梅袁伟成
遵义医科大学学报 2014年2期
白 玫,左 建,赵建强,张晓梅,袁伟成
(1.中国科学院 成都有机化学研究所,四川 成都 610041;2. 中国科学院大学,北京 100049;3. 遵义医学院 药学院, 贵州 遵义 563099)
氧化吲哚作为一类重要的结构单元广泛存在于天然产物中,尤其是3,3-二取代氧化吲哚更是一些天然产物和药物活性分子的核心骨架[1]。因此,深入开展有机合成方法学的研究,合成结构多样化的3,3-二取代氧化吲哚类化合物,近年来在有机合成化学以及药物化学研究领域受到广泛关注[2]。然而,在已报道的合成方法中,存在原料不易获得、反应条件苛刻、反应收率较低等局限性。因此,发展简单高效的合成3,3-二取代氧化吲哚类化合物的方法具有重要的意义。
3,3-二取代氧化吲哚类化合物的合成主要通过以下两种常规的反应实现:①亲核试剂对靛红或靛红亚胺的亲核加成反应[3-4];②3-单取代氧化吲哚对亲电试剂的加成反应[5-6]。近来,Stoltz课题组报道了丙二酸酯对3-卤代氧化吲哚的加成反应,证明了3-卤代氧化吲哚是一类很好的亲电试剂[7]。β-酮酸是一类重要的碳亲核试剂[8],肟是一类重要的氧亲核试剂[9],苯亚磺酸钠是一类重要的硫亲核试剂[10],此3类化合物作为亲核试剂参与的反应已有相关文献报道。鉴于此,如果此3类化合物分别能够与3-卤代氧化吲哚进行反应,就可实现C3位含C、O以及S原子的3,3-二取代氧化吲哚的合成。
在此,设计并合成了一类新的3-卤代氧化吲哚,在金鸡纳碱衍生的催化剂(Cat 1)与磷酸钾(K3PO4)的催化作用下,分别实现了3-卤代氧化吲哚与β-酮酸、肟以及苯亚磺酸钠的反应,高收率和高选择性的得到了一系列C3位含C、O以及S原子的3,3-二取代氧化吲哚类化合物。
1 实验与方法
1.1 材料 仪器:BrukerAV-300型核磁共振仪、Perkin-Elmer-341自动旋光仪、Büchi B-545熔点仪、LC-20AD高效液相色谱仪、手性色谱柱(Chiralpak AD)、旋转蒸发仪、磁力搅拌器等。试剂:苯甲醛、哌啶、2-吲哚酮、N-溴代丁二酰亚胺(NBS)、N-氯代丁二酰亚胺(NCS)、硼氢化钠等。
1.2 实验原理 在碱性条件下,3-卤代氧化吲哚失去一分子的卤化氢生成不饱和的亚胺中间体,亲核试剂对亚胺中间体进行1,4-加成反应,得到最终的3,3-二取代氧化吲哚类化合物(见图1)。
1.3 实验与步骤
1.3.1 3-卤代氧化吲哚(1a与1b)的合成(见图2)。
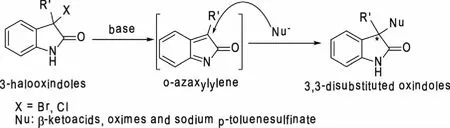
图1 亲核试剂对3-卤代氧化吲哚的加成反应
①称取2-吲哚酮A(1.33 g,10 mmol) 于50 mL的圆底烧瓶中,依次加入无水乙醇(30 mL),吡咯烷(0.1 mL),苯甲醛(1.06 g, 10 mmol),加热回流反应2 h,得化合物B的粗产品。②将化合物B的乙醇溶液冷却至0 ℃,加入NaBH4(1.9 g,50 mmol),在室温下搅拌2 h。待反应完全后,加水淬灭反应,乙酸乙酯萃取,合并有机相,无水硫酸钠干燥,减压浓缩得化合物C的粗产品。③将化合物C溶解在40 mL二氯甲烷后,加入0.3 mL三乙胺和NBS(或NCS) (1.77 g,10 mmol),并于室温搅拌反应1 h。待反应完全后,减压浓缩,残留物经柱层析(石油醚:乙酸乙酯 = 10∶1)得粗产品,乙酸乙酯与石油醚重结晶得化合物1a (或1b)。
Compound 1a:1H NMR(CDCl3,300 MHz)δ(ppm):3.68(d,J=13.5 Hz,1H),3.74(d,J=13.5 Hz,1H),6.75(d,J=7.8 Hz,1H),6.98-7.01(m,2H),7.06-7.13(m,4H),7.17-7.20(m,1H),7.38(d,J=7.5 Hz,1H),8.50(s,1H);13C NMR(75 MHz,DMSO-d6)δ(ppm):43.5,58.0,110.2,122.2,125.6,127.1,128.0,130.2,130.3,130.4,134.8,140.8,174.4.HRMS(ESI)Calcd.forC15H13BrNO[M+H]+:302.0175;Found:302.0172。
Compound 1b:1H NMR (300 MHz, CDCl3)δ(ppm): 3.56 (s, 2H), 6.76 (d,J= 7.8 Hz, 1H), 6.98-7.01 (m, 2H), 7.08-7.13 (m, 4H), 7.20-7.22 (m, 1H), 7.30 (d,J= 7.5 Hz, 1H), 8.42 (s, 1H);13C NMR (75 MHz, DMSO-d6)δ(ppm): 43.4, 66.1, 110.2, 122.2, 125.3, 127.2, 127.9, 128.4, 130.3, 133.9, 141.2, 174.0. HRMS (ESI) Calcd. for C15H12ClNNaO [M+Na]+: 280.0500; Found: 280.0506。
1.3.2 3-卤代氧化吲哚(1a与1b)与β-酮酸(5)的反应用于合成化合物6 化合物6的合成:于硬质反应管中加入3-卤代氧化吲哚1a(或1b) (0.1 mmol)、β-酮酸5(0.12 mmol)、Cat 1(0.02 mmol) 以及K3PO4(0.1 mmol),最后加入2 mL甲苯,混合物在室温下搅拌15 h,TLC跟踪反应进程。待反应进行完全后,混合物直接经柱层析分离得产物6。
Compound 6: Pale yellow soild, 31.0 mg, yield 91%; 90%ee,[α]D20=+112.3(c1.35,CHCl3);m.p.158.4-159.7 ℃.1H NMR (300 MHz, CDCl3)δ(ppm): 3.10 (d,J= 12.8 Hz, 1H), 3.19 (d,J= 12.8 Hz, 1H), 3.78 (d,J= 18.3 Hz, 1H), 3.84 (d,J= 18.3 Hz, 1H), 6.72 (d,J= 7.8 Hz, 1H), 6.90-6.99 (m, 4H), 7.09-7.13 (m, 4H), 7.41 (t,J= 7.5 Hz, 2H), 7.52 (d,J= 7.5 Hz, 1H), 7.86 (d,J= 7.2 Hz, 3H);13C NMR (75 MHz, CDCl3)δ(ppm): 44.0, 44.7, 51.1, 109.8, 121.3, 123.0, 126.6, 127.5, 127.7, 127.8, 128.3, 130.2, 131.3, 133.0, 134.9, 136.2, 141.6, 181.7, 195.9. HRMS (ESI-TOF) Calcd. for C23H19NNaO2[M+Na]+: 364.1308; Found: 364.1318. HPLC analysis: Chiralcel AD-H column,i-propanol/hexane = 50:50, flow rate 1.0 mL/min,λ= 254 nm,tmajor= 16.5 min,tminor= 7.4 min。
1.3.3 3-卤代氧化吲哚(1a与1b)与肟(2)的反应用于合成化合物3以及化合物4 化合物3的合成:于硬质反应管中加入3-卤代氧化吲哚1a(或1b) (0.1 mmol),肟2(0.12 mmol),Cat 1(0.02 mmol) 以及K2CO3(0.08 mmol),最后加入1 mL四氢呋喃,混合物在室温下搅拌12 h,TLC跟踪反应进程。待反应完全后,混合物直接经柱层析分离得产物3。
Compound 3: White solid, yield 89%; 99% ee, [α]D20=+63.9 (c 3.50, CHCl3); m.p. 59.5-61.7oC.1H NMR (300 MHz, DMSO-d6),δ(ppm): 3.19 (d, J = 12.6 Hz, 1H), 3.41 (d,J= 12.6 Hz, 1H), 6.64 (d,J= 7.5 Hz, 1H), 6.87-6.97 (m, 3H), 7.10-7.15 (m, 4H), 7.19 (d,J= 7.5 Hz, 1H), 7.35-7.42 (m, 5H), 8.32 (s, 1H), 10.33 (s, 1H);13C NMR (300 MHz, DMSO-d6)δ(ppm): 40.1, 85.6, 109.6, 121.5, 125.0, 126.8, 127.0, 127.8, 128.2, 128.9, 129.7, 130.4, 130.5, 131.4, 133.8, 142.5, 150.4, 175.8. HRMS (ESI) Calcd. for C22H18N2NaO2[M+Na]+: 365.1260; Found: 365.1257. HPLC conditions: Chiralcel AD-H column, EtOH/hexane 30:70, flow rate 1.0 mL/min,λ= 254 nm,tmajor= 20.22 min,tminor= 9.27 min。
化合物4的合成:于硬质反应管中加入3 (34.2 mg, 0.1 mmol),加入5 mL无水甲醇及10 %的钯炭(21 mg),通氢气(1.0 atm),室温下搅拌反应20 h。反应完毕后,混合物经硅藻土过滤,真空除去溶剂得残留物,残留物经柱层析柱色谱分离得产物4。
Compound 4: White solid, yield 90 %; 98 % ee, [α]D20=+17.6 (c 2.80, MeOH); m.p. 168.1-170.5oC.1H NMR (300 MHz, DMSO-d6)δ(ppm): 2.99 (d,J= 12.6 Hz, 1H), 3.15 (d,J= 12.6 Hz, 1H), 6.12 (s, 1H), 6.59 (d,J= 7.8 Hz, 1H), 6.87-6.92 (m, 3H), 7.07-7.12 (m, 5H), 10.05 (s, 1H);13C NMR (300 MHz, DMSO-d6)δ(ppm): 43.6, 76.7, 109.4, 121.3, 124.7, 126.4, 127.6, 129.0, 130.2, 131.0, 135.1, 141.7, 178.9. HRMS (ESI) Calcd. for C15H13NNaO2[M+H]+: 262.0838; Found: 262.0839. HPLC conditions: Chiralcel AD-H column,i-PrOH/hexane = 30:70, flow rate 1.0 mL/min,λ= 254 nm,tmajor= 5.69 min,tminor= 6.56 min。
1.3.4 3-卤代氧化吲哚(1a与1b)与对甲基苯亚磺酸钠(7)的反应用于合成化合物8 化合物8的合成:于硬质反应管中加入1a(或1b) (0.1 mmol),对甲基苯亚磺酸钠7(0.12 mmol) 以及K3PO4(0.02 mmol),然后加入2 mL乙腈,混合物在室温下搅拌12 h,TLC跟踪反应进程。待反应进行完全后,减压蒸掉乙腈,残留物经柱层析分离得产物8。
Compound 8: White solid, 98% yield; m.p. 268.4-270.1oC.1H NMR (300 MHz, DMSO-d6)δ(ppm): 2.35 (s, 3H), 3.59 (d,J= 12.9 Hz, 1H), 3.67 (d,J= 12.9 Hz, 1H), 6.47 (d,J= 7.5 Hz, 1H), 6.93-7.10 (m, 6H), 7.17 (t,J= 7.5 Hz, 1H), 7.32 (d,J= 8.1 Hz, 2H), 7.41 (d,J= 7.8 Hz, 2H), 7.67 (d,J= 7.2 Hz, 1H), 10.48 (s, 1H);13C NMR (75 MHz, DMSO-d6)δ(ppm): 21.1, 34.4, 75.8, 109.5, 121.6, 121.7, 127.0, 127.1, 128.0, 129.3, 130.0, 130.4, 131.9, 133.4, 142.9, 145.3, 170.4. HRMS (ESI-TOF) calcd for C22H19NNaO3S [M + Na]+: 400.0978; found: 400.0974。
2 结果
从图3可以看出,在碱以及Cat 1的催化下,3-卤代氧化吲哚1a和1b可以分别与碳亲核试剂β-酮酸、氧亲核试剂苯甲醛的肟、以及硫亲核试剂对甲苯亚磺酸钠反应,高收率和高选择性的得到一系列C3位含有C、O以及S原子的3,3-二取代氧化吲哚类化合物。值得一提的是,3-卤代氧化吲哚1a和1b与苯甲醛的肟反应的产物3经一步简单的还原即可得到四取代3-羟基氧化吲哚类化合物4,该结构单元是一些药物活性分子的核心骨架[11]。
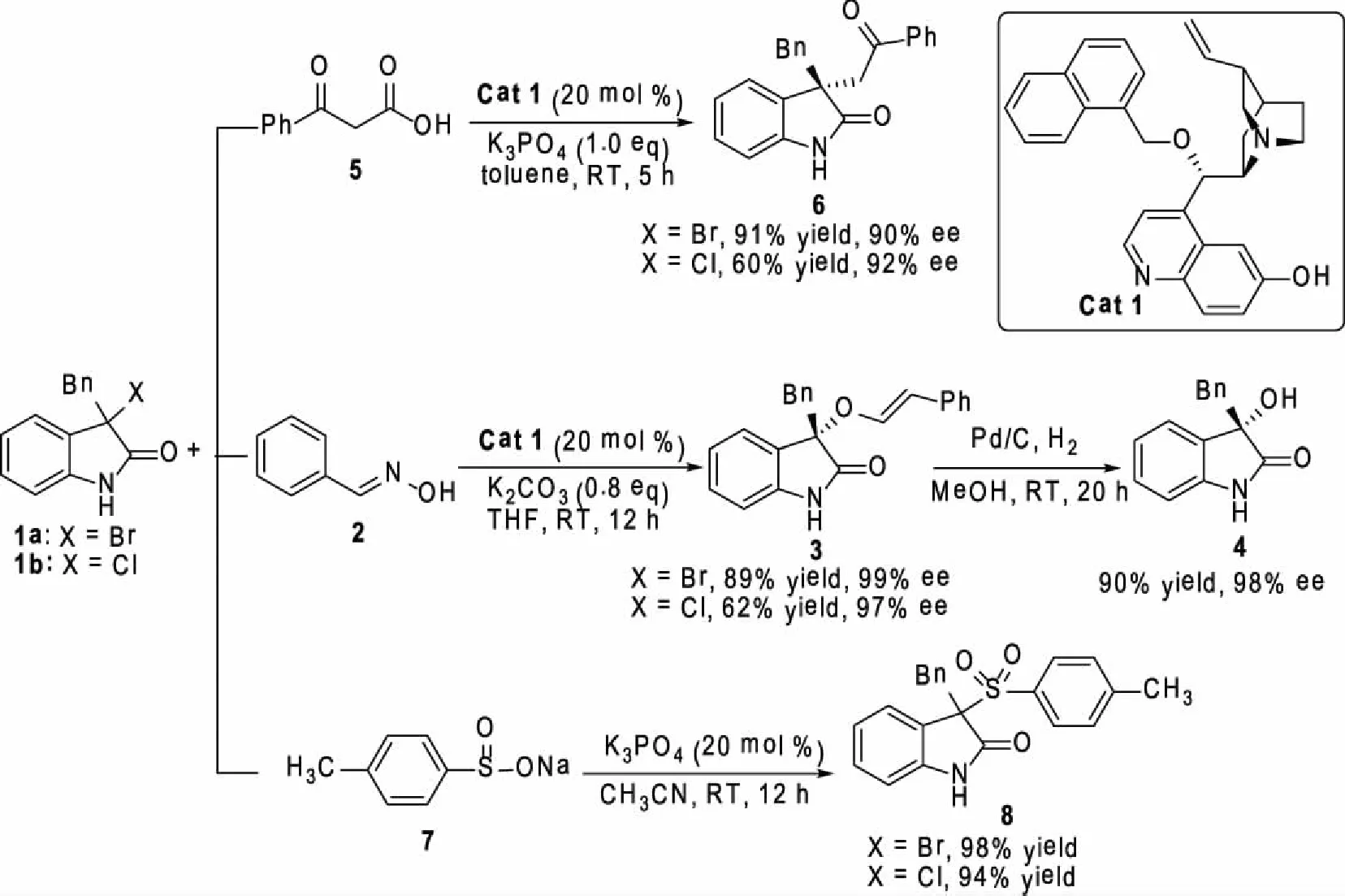
图3 3,3-二取代氧化吲哚的合成
3 讨论
以商业易得的2-吲哚酮为原料经3步反应合成了3-卤代氧化吲哚1a和1b,并成功实现了3-卤代氧化吲哚与β-酮酸、肟以及对甲苯亚磺酸钠的加成反应。该方法具有原料及催化剂易得,反应条件温和,操作简单等特点,为合成C3位含C、O以及S原子的3,3-二取代氧化吲哚类化合物提供了一条有效的途径。
[参考文献]
[1] Trost B M,Brennan M K.Asymmetric syntheses of oxindole and Indole spirocyclic alkaloid natural products[J].Synthesis,2009,40(15):3003-3025.
[2] Singh G S, Desta Z Y. Isatins as privileged molecules in design and synthesis of spiro-fused cyclic frameworks[J].Chem. Rev, 2012, 112(11): 6104-6155.
[3] Hanhan N V, Ball-Jones N R, Tran N T,et al.Catalytic asymmetric[3+2]annulation of allylsilanes with Isatins:synthesis of spirooxindoles[J].Angew chem Int Ed Enql,2012,51(4):989-992.
[4] Guo Q X, Liu Y W, Li X C,et al.Enantioselective and solvent-controlled diastereoselective mannich reaction of isatin imines with hydroxyacetone:synthesis of 3-substituted 3-aminooxindoles[J].J Org Chem, 2012, 77(7): 3589-3594.
[5] Cui B D, Han W Y, Wu Z J,et al.Enantioselective synthesis of quaternary 3-aminooxindoles via organocatalytic asymmetric michael addition of 3-monosubstituted 3-aminooxindoles to nitroolefins[J].J Org Chem,2013,78(17):8833-8839.
[6] Tian X,Jiang K,Peng J,et al.Organocatalytic stereoselective mannich reaction of 3-substituted oxindoles[J].Org Lett,2008,10(16):3583-3586.
[7] Krishnan S,Stoltz B. M.Preparation of 3,3-disubstituted oxindoles by addition of malonates to 3-halo-3-oxindoles[J]. Tetrahedron Lett,2008,48(43): 7571-7573.
[8] Evans D. A, Mito S, Seidel D. Scope and mechanism of enantioselective michael additions of 1,3-dicarbonyl compounds to nmitroalkenes catalyzed by nickel(II)-diamine complexes[J].J Am Chem Soc,2007, 129(37): 11583-11592.
[9] Zhang F G, Yang Q Q, Xuan J,et al. Enantioselective conjugate addition of oximes to trisubstituted β-Nitroacrylates:an organocatalytic approach to β2,2-amino acid derivatives[J].Org Lett, 2010, 12(24): 5636-5639.
[10] Procter D J. The synthesis of thiols, selenols, sulfides, selenides, sulfoxides, selenoxides, sulfones and selenones[J]. J Chem Soc, 2001,1(4):335-354.
[11] Zhou F, Liu Y L, Zhou J. Catalytic asymmetric synthesis of oxindoles bearing a tetrasubstituted stereocenter at the C-3 position[J].Adv Synth Catal, 2010, 352(9): 1381-1407.
猜你喜欢
水产科学(2021年3期)2021-05-24
中国造纸(2020年12期)2021-01-08
中华养生保健(2020年2期)2020-11-16
云南医药(2020年5期)2020-10-27
医学新知(2019年4期)2020-01-02
吉林农业(2019年6期)2019-06-11
教育教学论坛(2018年38期)2018-09-25
中国实用医药(2018年8期)2018-03-27
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
