
查尔酮类化合物的抗肿瘤活性及其构效关系研究进展
2014-12-03 03:07陈建忠聊城市人民医院药学部山东聊城252000
中国药房 2014年5期
陈建忠(聊城市人民医院药学部,山东聊城 252000)
查尔酮是广泛存在于甘草、啤酒花、镰形棘豆等植物中的一类天然化合物,其基本母核为1,3-二苯基丙烯酮或苯基苯乙烯基酮,具有多种药理活性,并作为一种天然的有机颜料被广泛应用于日常生产中。
在结构方面,由于查尔酮类化合物分子具有较大的柔性,能与不同的受体结合,因而拥有广泛的生物活性[1-2],如抗肿瘤、抗炎、抗真菌、抗病毒、治疗糖尿病等。此外,α、β烯酮结构是软亲电试剂,并不与脱氧核糖核酸(DNA)或核糖核酸(RNA)结构中的硬亲核试剂如氨基、羟基发生反应,从而防止了突变或癌症的发生。de Vincenazo R等[3]和Shibata S[4]分别提出查尔酮类化合物对卵巢癌和胃癌均有明显的抑制作用。近年来,国内外的科学工作者对查尔酮类化合物的生物活性及其结构修饰进行了广泛而深入的研究。本文重点对PubMed数据库中1994-2013年查尔酮类化合物的抗肿瘤活性及其构效关系的研究进展进行阐述。
1 查尔酮类化合物的抗肿瘤活性研究
据报道,查尔酮类化合物可通过诱导肿瘤细胞凋亡和阻滞肿瘤细胞分裂而抑制肿瘤细胞增殖,同时使p53蛋白表达增加,细胞周期素A、B以及细胞周期蛋白依赖性激酶1(CDK1)分子的表达减少[5]。也有学者认为,查尔酮是通过作用于双微小体2(MDM2)影响p21蛋白转录而达到抗肿瘤细胞增殖的作用。此外,查尔酮类化合物还能通过抑制蛋白酶的降解来发挥其抗肿瘤作用[6]。
为了系统评价查尔酮类化合物的抗肿瘤活性,根据其母核上有效取代基类型的不同,可将其分为羟基化和甲基化查尔酮、卤取代查尔酮、硼酸基取代查尔酮和含氮基团取代查尔酮。不同取代基取代的查尔酮类化合物见表1。
1.1 羟基化和甲基化查尔酮
具有较高抗肿瘤活性的查尔酮类化合物多数都含有羟基和甲基结构,如康普立停A-4和秋水仙碱就是最具代表性的化合物。康普立停A-4是一种小分子肿瘤血管破坏药物[7],能明显减少头颈部癌、食管癌、肺癌等多种肿瘤血管的血流量,使肿瘤组织坏死,但对正常组织的血管不起作用,具有特异性和低毒性,目前我国研发的该类新药已进入Ⅱ期临床试验阶段。秋水仙碱[8]是从百合科植物秋水仙中提取出的化合物,对乳腺癌具有较好的疗效,能抑制乳腺癌细胞纺锤体的形成,并与微管蛋白二聚体结合,阻止微管蛋白转换,使肿瘤细胞停滞于有丝分裂中期,从而导致肿瘤细胞凋亡。Devi MA等[9]认为,当体内出现大量的恶性淋巴细胞时,一些天然的多酚羟基查尔酮类化合物(如化合物1)通过自分泌以及旁分泌途径促进白细胞介素2(IL-2)生成,抑制恶性淋巴细胞的增殖。
Lawrence NJ等[10]合成出了20种羟基和甲基取代位置不同的查尔酮类化合物,其中4′-羟基-2,3′,4,6′-四甲基查尔酮1对慢性髓细胞白血病细胞K562的半数抑制浓度(IC50)达30 nmol/L。Rao YK等[11]提出,2′-OH是化合物保持抗肿瘤活性必不可少的取代基团,其中化合物2是活性最好也是研究最多的一类,其对组织细胞淋巴瘤细胞U937表现出明显的增殖抑制作用,但其作用机制尚未见报道。Zi X等[12]从一种胡椒科类植物卡瓦根中提取出了一类含有三甲基取代A环的查尔酮类化合物Flawkawain。该化合物通过凋亡相关基因Bax蛋白依赖途径以及线粒体依赖途径诱导膀胱癌细胞凋亡,并且对荷瘤小鼠的肿瘤组织也具有明显的抑制作用。Boumendjel A等[13]合成出了一种羟基和甲基取代的新型查尔酮类化合物,其在5 μg/ml时能阻滞K562细胞于G2/M期,表现出了较好的体外抗肿瘤活性。
Yamazaki S等[14]从甘草中提取出了一种新型查尔酮类化合物异甘草素。研究发现,该化合物能抑制结肠癌HCT116细胞的增殖及转移,也可诱导乳腺癌、胃癌、肝癌、黑色素瘤等肿瘤细胞凋亡。Saxena HO等[15]证实,查尔酮类化合物能使肺癌A549细胞周期停滞于G1期或G2/M期,同时也能促进这两个时期的细胞凋亡;此外,该化合物还能促进p53、p21蛋白以及细胞凋亡抑制因子(Fas/APO-1)和Fas配位体的表达,进而诱导细胞凋亡。Hsu YL等[5]报道,异甘草素也能诱导肝癌细胞株HepG2细胞凋亡和细胞周期阻滞,这与p53、p21、Fas/APO-1受体、Fas配位体、Bax蛋白和Noxa等调控因子有关,但又同时指出异甘草素的抗肿瘤作用与凋亡前体蛋白降解无关。Kanazawa M等[16]研究发现,异甘草素对前列腺癌细胞系DU145和LNCaP均具有明显的细胞毒作用,可将细胞阻滞于S、G2/M期,增强了GADD153 mRNA和蛋白的表达,并且能呈剂量依赖性地促进GADD153启动子的活性。
Kahyo T等[17]合成出了2′,4′-二羟基-6′-甲氧基-3′,5′-二甲基查尔酮(化合物4)。该化合物能显著抑制人白血病细胞K562增殖。而合成的另一类新型化合物3,2,3,4-四甲基查尔酮能促进p21蛋白表达的增加以及p53蛋白的高度乙酰化,并且作为一种新型的长效蛋白质去乙酰化酶(Sirtuin type 1,SIRT1)抑制剂抑制SIRT1的脱乙酰化来发挥其抗肿瘤作用。
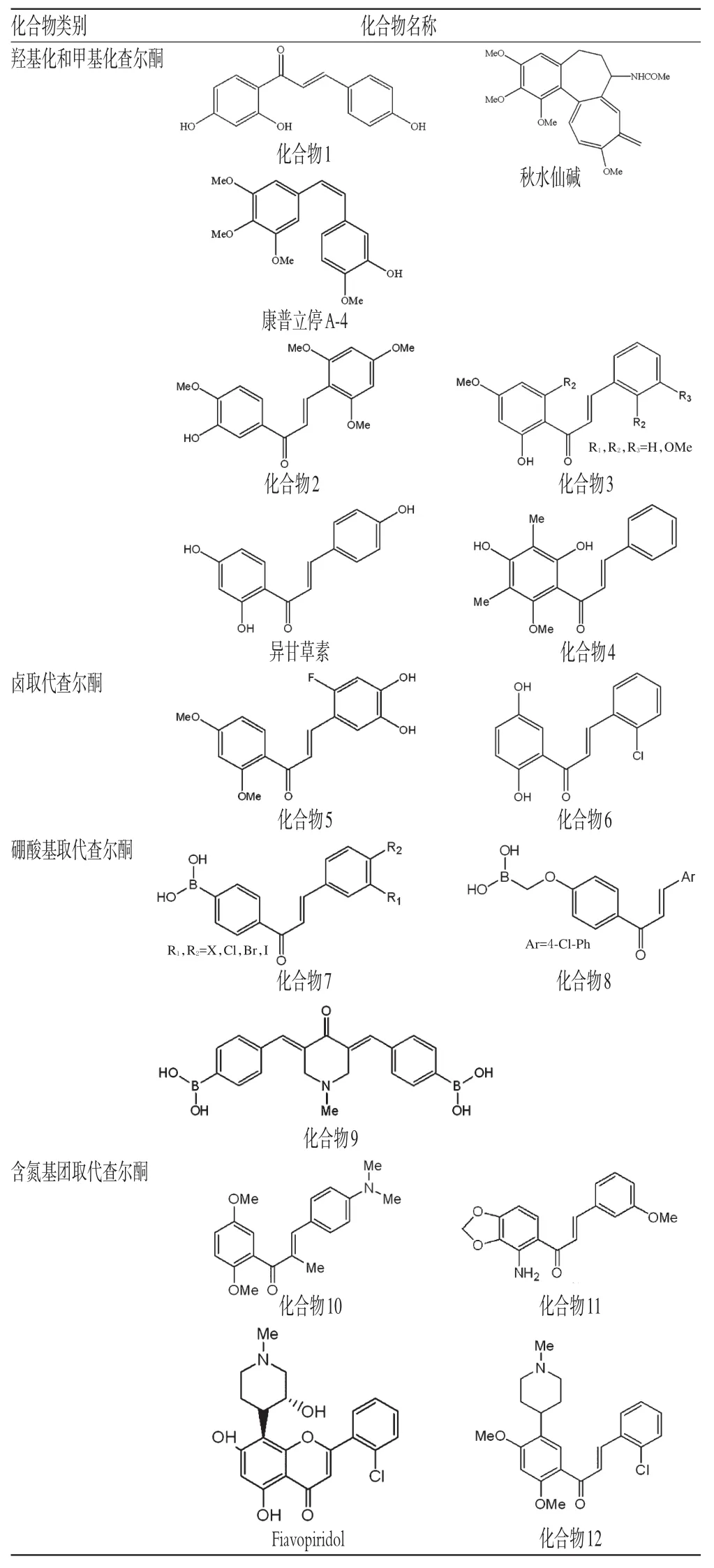
表1 不同取代基取代的查尔酮类化合物
1.2 卤取代查尔酮
近年来,采用卤取代是合成查尔酮类化合物的另一个新途径。Nakamura C等[18]第一次合成出了氟取代的查尔酮类化合物。将C-6引入氟原子得到3,4-二羟基-2′,4′-二甲氧基-6-氟查尔酮(化合物5)。该化合物的抗肿瘤活性可与经典抗肿瘤药长春新碱相比,且当腹腔注射(ip)于荷瘤小鼠模型时,抑瘤率更是达到了60%以上。Nam NH等[19]验证了A环上卤素、羟基、甲氧基取代都会使查尔酮的抗肿瘤活性增加,其中最有效的化合物是2-氯-2′,5′-二羟基查尔酮(化合物6),其在低浓度时的抑制率达到了60%以上。
1.3 硼酸基取代查尔酮
硼酸基取代查尔酮作为一种荧光探针用来检测糖类化合物的存在,而Johns Hopkins医院的专家合成出了一系列硼酸基取代查尔酮类化合物(化合物7)。经体外试验表明,该类化合物能高度选择性地、特异性地抑制乳腺癌细胞的增殖。Kumar SK等[20]提出,硼酸基取代查尔酮的抗肿瘤作用与一些大分子蛋白有密切的联系,该类化合物能分解MDM2/p53蛋白复合物,并且显著增加p53和p21蛋白的堆积。Modzelewska A等[21]也指出,硼酸基取代的查尔酮类化合物对乳腺癌细胞具有很高的特异性,而在原有的硼酸基取代的基础上加入一系列的芳香基团(化合物8)后更能增强抗肿瘤疗效。
Achanta G等[6]合成出了一系列双查尔酮类化合物,其中3,5-二-(4-硼酸基-苯甲基)-1-甲基哌啶-4-酮化合物(化合物9)在MTT试验中作用于结肠癌细胞系SW620、HT-29,其IC50分别为1.5 μmol/L和0.6 μmol/L。在MDM2/p53蛋白复合物复合体未分解的状态下,此类双查尔酮类化合物可促进p53和p21蛋白的堆积,尤其在放疗的情况下可优先杀死p53+/+细胞株。
Buolamwini JK等[22]认为,硼酸基取代的查尔酮有两种可能的抗肿瘤作用机制:MDM2/p53复合体的降解和蛋白酶体抑制。由此说明,硼酸基取代的查尔酮是一种具有较大潜力的抗肿瘤化合物。
1.4 含氮基团取代查尔酮
为了改善查尔酮类化合物的理化性质及药动学活性,许多科学家的研究重点偏向于在查尔酮母核结构中引入含氮杂环。抗肿瘤化合物MDL27048(化合物10)是第1个合成的含氮基团取代的查尔酮,它作用于微管蛋白,并且通过细胞周期阻滞抑制肿瘤细胞的增殖。Xia Y等[23]研究发现,2′-含氮基团是抗肿瘤作用的有效基团。通过体外筛选,6′-氨基-3-甲氧基-4′,5′-亚甲基二氧查尔酮(化合物11)是其中抗肿瘤作用最强的化合物。文献也报道了含氮的黄酮类似物对CDK1/cyclin B具有抑制作用[24]。
利用已知抗肿瘤药Flavopiridol的细胞周期依赖性蛋白激酶(CDKs)为先导化合物设计出的一类哌啶基团取代的查尔酮化合物,其B环上有哌啶环取代,且N原子上连有甲基。其中,2-氯-2′,4′-二甲氧基-5-N-甲基哌啶查尔酮(化合物12)的抗肿瘤效果要明显强于先导化合物CDKs。该化合物主要通过阻滞MCF-7细胞和HCT116细胞的G0/G1和G2/M期的正常分裂以及下调CDK4、细胞周期素B、E2F等调控因子来抑制肿瘤细胞增殖。
Meng CQ等[25]发现了一类新型杂环芳基取代的查尔酮类化合物,该类化合物能抑制肿瘤坏死因子α(TNF-α)诱导的血管分子黏附分子1(VCAM-1)的表达,从而达到杀灭肿瘤组织的效果。
2 构效关系研究
药物化学家从查尔酮的基本骨架出发,在A、B环不同位置引入不同的取代基团,逐步形成了一个查尔酮类化合物的分子库,并且通过筛选得到了一系列具有较好抗肿瘤活性的化合物。Kelland LR[26]发现,A环上不同位置取代的2′,5′-二羟基查尔酮的细胞毒性有明显的不同,当在A环的2、4、6位置上引入卤素、羟基、甲氧基等吸电子基团时查尔酮的抗肿瘤活性明显增加,最具代表性的化合物是2-氯-2′,5′-二羟基查尔酮(化合物6),该化合物在体外和体内都有明显的抗肿瘤活性。A环的多甲氧基结构是一个优良的药效基团,当在3、4、5位引入三甲氧基结构时,该化合物能显著抑制微管蛋白聚合活性。此外,引入α甲基、芳基及2′-含氧基团均可增强化合物的抗有丝分裂活性。引入Mannich基团是增强查尔酮类化合物对肿瘤细胞毒活性的一类有效途径,并且引入位置在A环时比B环抗肿瘤效果更好,如化合物7、化合物8、化合物9是一类含硼查尔酮类化合物,该类化合物对MCF-7细胞具有显著的细胞毒性。
在B环2′,4′,6′位置上引入并表现出抗肿瘤活性的取代基有酚羟基、羧基、甲氧基、乙氧基、二甲氨基、二乙氨基、异丙基等,其中最典型的是抗肿瘤药Flavopiridol[6]。Jun N等[27]合成了一系列含多元羟基取代的查尔酮类化合物,其中在骨架结构的2′,4′,6′-位上引入三羟基的化合物蛋白酪氨酸激酶(PTK)抑制作用最明显,而B环部分的邻苯二酚结构对抗肿瘤活性无显著影响。A环3,4,5-三甲氧基的查尔酮类化合物在B环引入硼酸基后明显增强了对肿瘤的抑制作用,但是其抑制微管蛋白聚合活性几乎消失。可见,不同基团的相互作用也是开发查尔酮类抗肿瘤药时应该关注之处。
丙烯酮的结构也会影响化合物的抗肿瘤活性,当碳-碳双键上的氢被甲氧基、氨基、氯取代时,可减弱肿瘤细胞的有丝分裂,抑制细胞增殖[28]。
反式构型的α、β双键在天然提取的以及后期合成的查尔酮类化合物中广泛存在,且大部分都具有显著的抗肿瘤活性。当A环引入α甲基时,可将查尔酮类化合物锁定在反式构象中,使其活性增加约40倍[29]。而顺式的α、β双键查尔酮类化合物(以抗肿瘤新药康普立停A-4为代表)也逐渐引起了人们的广泛关注[7]。
3 结语
如今,癌症已成为威胁人类健康的常见病和多发病,因此研制安全、有效的抗肿瘤药对人类的生存和发展显得尤为重要。近十多年来,众多学者围绕查尔酮进行了大量的结构修饰工作,以期获得高效、低毒的查尔酮类化合物。研究表明,查尔酮类化合物药理活性广泛、安全,且结构简单、制备方便,是一种良好的药物开发先导物。
经过十多年的开发,目前已有多个查尔酮类药上市,如索法酮和美托查酮。该类化合物的抗肿瘤机制大致可分为以下几个方面:(1)阻滞细胞周期;(2)促进肿瘤细胞凋亡;(3)抑制肿瘤血管生成;(4)促进抑癌基因的表达;(5)蛋白酪氨酸激酶(PTK)表达的增加,从而抑制细胞信号的转导;(6)诱导淋巴细胞对IL-2的分泌等。因此,进一步研究查尔酮类化合物的结构、药理活性以及毒性的关系,对开发查尔酮类抗肿瘤药是一项十分重要和有价值的工作,抗肿瘤新药的研究也将具有广阔的前景。
[1]陈季武,胡天喜,朱大元.11种黄铜化合物清除超氧阴离子的构效关系研究[J].中国药学杂志,2004,37(1):57.
[2]廖头根,汪秋安,方伟琴,等.新型查尔酮类化合物的合成及其生物活性研究[J].有机化学,2006,26(5):685.
[3]de Vincenazo R,Ferlini C,Distefano M,et al.In vitro evaluation of newly developed chalcone analogues in human cancer cells[J].Cancer Chemother Pharmacol,2000,46(4):305.
[4]Shibata S.Antitumor igenic chalcone[J].Stem Cell,1994,12(1):44.
[5]Hsu YL,Kuo PL,Lin CC.Isoliquiritigenin induces apoptosis and cell cycle arrest through p53-dependent pathway in HepG2 cells[J].Life Sci,2005,77(3):279.
[6]Achanta G,Modzelewska A,Feng L,et al.A boronicchalcone derivative exhibits potent anticancer activity through inhibition of the proteasome[J].Mol Pharmacol,2006,70(1):426.
[7]Bruce CB.Antivascular therapy of cancer:DMXAA[J].Lancet Oncol,2003,4(3):141.
[8]Nguyen TL,Mcgrath C,Hermone AR,et al.A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach[J].J Med Chem,2005,48(19):6017.
[9]Devi MA,Das NP.In vitro effects of natural plant polyphenols on the proliferation of normal and abnormal human lymphocytes and their secretions of interleukin-2[J].Cancer Lett,1993,69(3):191.
[10]Lawrence NJ,Rennison D,McGown AT,et al.Linked parallel synthesis and MTT bioassay screening of substituted chalcones[J].J Comb Chem,2001,3(5):421.
[11]Rao YK,Fang SH,Tzeng YM.Differential effects of synthesized 2′-oxygenated chalcone derivatives:modulation of human cell cycle phase distribution[J].Bioorg Med Chem,2004,12(10):2679.
[12]Zi X,Simoneau AR.Flavokawain A.A novel chalcone from kava extract,induces apoptosis in bladder cancer cells by involvement of bax protein-dependent and mitochondria-dependent apoptotic pathway and suppresses tumor growth in mice[J].Cancer Res,2005,65(8):3479.
[13]Boumendjel A,Sotoing TG,Ngo BE,et al.Occurrence of the synthetic analgesic tramadol in an African medicinal plant[J].Angew Chem Int Ed Engl,2013,52(45):11780.
[14]Yamazaki S,Morita T,Endo H,et al.Isoliquiritigenin suppresses pulmonary metastasis of mouse renal cell carcinoma[J].Cancer Lett,2002,183(1):23.
[15]Saxena HO,Faridi U,Kumar JK,et al.Synthesis of chalcone derivatives on steroidal framework and their anticancer activities[J].Steroids,2007,72(13):892.
[16]Kanazawa M,Satomi Y,Mizutani Y,et al.Isoliquiritigenin inhibits the growth of prostate cancer[J].Eur Urol,2003,43(5):580.
[17]Kahyo T,Ichikawa S,Hatanaka T,et al.A novel chalcone polyphenol inhibits the deacetylase activity of SIRT1 and cell growth in HEK293T cells[J].J Pharmacol Sci,2008,108(3):364.
[18]Nakamura C,Kawasaki N,Miyataka H,et al.Synthesis and biological activities of fluorinated chalcone derivatives[J].Bioorg Med Chem Lett,2002,12(3):699.
[19]Nam NH,Kim Y,You Y,et al.Cytotoxic 2′,5′-dihydroxychalcones with unexpected antiangiogenic activity[J].Eur J Med Chem,2003,38(2):179.
[20]Kumar SK,Hager E,Pettit C,et al.Design,synthesis,and evaluation of novel boronic-chalcone derivatives as antitumor agents[J].J Med Chem,2003,46(14):2813.
[21]Modzelewska A,Pettit C,Achanta G,et al.Anticancer activities of novel chalcone and bis-chalcone derivative[J].Bioorg Med Chem,2006,14(10):3491.
[22]Buolamwini JK,Addo J,Kamath S,et al.Small molecule antagonists of the MDM2 oncoprotein as anticancer agents[J].Curr Cancer Drug Targets,2006,5(1):57.
[23]Xia Y,Yang ZY,Xia P,et al.Antitumor agents.Part 202:novel 2′-amino chalcones:design,synthesis and biological evaluation[J].Bioorg Med Chem Lett,2000,10(8):699.
[24]Zhang SX,Ma JG,Bao YM,et al.Nitrogen-containing flavonoid analogues as CDK1/cyclin B inhibitors:synthesis,SAR analysis,and biological activity[J].Bioorg Med Chem,2008,16(15):7127.
[25]Meng CQ,Zheng XS,Ni L,et al.Discovery of novel heteroaryl-substituted chalcones as inhibitors of TNF-α-induced VCAM-1 expression[J].Bioorg Med Chem Lett,2004,14(6):1513.
[26]Kelland LR.Farnesyl transferase inhibitors in the treatment of breast cancer[J].Expert Opin Investig Drugs,2003,12(3):413.
[27]Jun N,Gao H,Jun K.Synthesis and evaluation of 2′,4′,6′-trihydroxychalcones as a new class of tyrosinase inhibitors[J].Bioorg Med Chem,2007,15(6):2396.
[28]Cabrera M,Simoens M,Falchi G,et al.Synthetic chalcones,flavanones,and flavones as antitumoral agents:biological evaluation and structure-activity relationships[J].Bioorg Med Chem,2007,15(10):3356.
[29]Sikander M,Malik S,Yadav D,et al.Cytoprotective activity of a trans-chalcone against hydrogen peroxide induced toxicity in hepatocellular carcinoma(HepG2) cells[J].Asian Pac J Cancer Prev,2011,12(10):2513.
猜你喜欢
农业与技术(2021年3期)2021-12-06
世界科学技术-中医药现代化(2021年7期)2021-11-04
世界科学技术-中医药现代化(2021年12期)2021-04-19
农药科学与管理(2019年8期)2019-11-23
天然产物研究与开发(2018年9期)2018-10-08
天然产物研究与开发(2018年7期)2018-08-21
天然产物研究与开发(2018年5期)2018-06-13
中成药(2017年4期)2017-05-17
中成药(2017年3期)2017-05-17
小猕猴学习画刊(2016年9期)2016-05-14
