
内嵌乙硅烷的推拉型有机分子的二阶非线性光学性质的理论研究
2015-01-04 06:10黄德林高艳蓉余盛萍陈彬彬杨明理
成都工业学院学报 2015年2期
黄德林,高艳蓉,余盛萍,* ,陈彬彬,杨明理
(1.西南民族大学 化学与环境保护工程学院,成都 610041;2.四川大学 a.化学工程学院;b.原子与分子物理研究所,成都 610065)
无机非线性光学(Nonlinear optical,NLO)材料广泛应用于激光技术、光学通信、数据储存及光信号处理等领域[1-2]。与无机材料相比,有机NLO材料具有响应系数大、响应时间短、易于设计和制备加工等优点[3]。但是热稳定性差、透光性差等缺陷限制了有机NLO材料的研发,至今只有极个别的有机NLO材料产品获得应用。常见的有机NLO材料分子具有给体(D)-共轭桥(π)-受体(A)结构。一方面,体系中的电子,尤其是共轭电子,受到两端供电子和吸电子基团的推拉作用,表现出较大的非线性极化响应。另一方面,共轭体系的存在常导致在可见光区形成较强的电子吸收带,因此这类化合物通常是不透明的。此外,由于较强的电子推拉作用,这类化合物分子常具有较大的基态偶极矩,在结晶过程中会受到较强的偶极-偶极相互作用的影响而交替地反向排列,形成具有对称中心的晶体结构,不再具有二阶NLO响应。为解决透光性和晶体工程上的困难,人们尝试设计出各种具有特殊结构的分子,如V型、H型和八极分子等[4-5]。最近,Shimada等[6]设计并制备了系列含乙硅烷的推拉型有机化合物,发现它们具有良好的透光性,其中个别样品具有较大的二阶NLO响应系数,但对其电子结构和NLO响应之间的关系缺乏研究。本文在研究该类化合物分子结构的基础上,分析它们具有良好透光性以及NLO响应系数之间产生差异的原因。最后,对该类化合物进行分子设计,在保持透光性的前提下,设计出具有更好NLO响应的该类化合物分子结构。
1 计算方法
各分子结构均在 B3LYP[7]/6-311++G(d,p)水平下进行结构优化,并计算其振动频率以确定其为稳定构型,得到分子的几何结构参数。采用TDDFT方法[8-9]在相同水平下计算各稳定构型的电子吸收光谱,得到第一吸收波长。采用相同的泛函和基组,利用有限场(FF)方法[10-11],计算分子的极化率和超极化率。计算使用Gaussian09程序完成[12]。
2 结果与讨论
图1为本文计算研究的分子结构示意图,其中1~5来自Shimada等[6]的实验研究。这些分子具有典型的D-π-A结构,但在2个苯环之间嵌入了含有饱和Si-Si键的乙硅烷,阻碍了苯环之间的共轭效应。表1列出了这些分子的部分结构参数。2个苯环中的C-C平均键长为0.139 6~0.140 5 nm,居于C-C单键及C=C双键的键长之间。Si-Si的键长约为0.237 nm,键级接近1,与在相同水平下计算得到的乙硅烷中的Si-Si键长0.236 nm相近,是典型的单键。计算表明:在各取代基及不同取代位置下,分子保持了共轭体系的特征,但被饱和的Si-Si单键分成了2个部分。
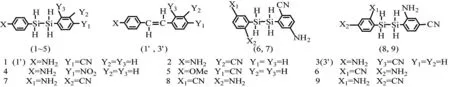
图1 推拉型有机分子的结构式
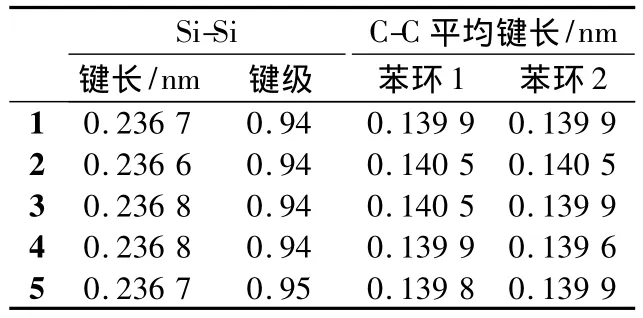
表1 内嵌乙硅烷的推拉型有机分子的部分几何结构参数
图2给出了TDDFT计算得到的电子吸收光谱。除4以外,其余4个分子的第一吸收峰都位于紫外区,且低于330 nm,这与实验观测一致。因此,这些化合物在较宽的波长范围内具有很好的透光性。在4中,硝基中的N原子和2个O原子与苯环形成共轭结构,扩大了共轭体系,降低了电子跃迁能,导致电子吸收光谱红移。上述各分子的第一电子吸收峰均由从最高占据轨道(HOMO)到最低空轨道(LUMO)的跃迁产生。图3给出了3的HOMO和LUMO构成图,前者主要由连接氨基的苯环的2p轨道和Si的3s轨道贡献,后者主要由连接氰基的苯环的2p轨道和Si的3s轨道贡献。这一过程需要克服Si-Si键的障碍,需要更高的能量,因此上述分子的第一吸收峰主要分布在紫外区。其余4个分子的HOMO和LUMO具有相似的构成。将饱和Si-Si键更换为不饱和的C=C双键,第一吸收峰发生较为明显的红移,如对应1和3的1'和3'的第一吸收峰分别红移了约50 nm和40 nm,已接近可见光区。
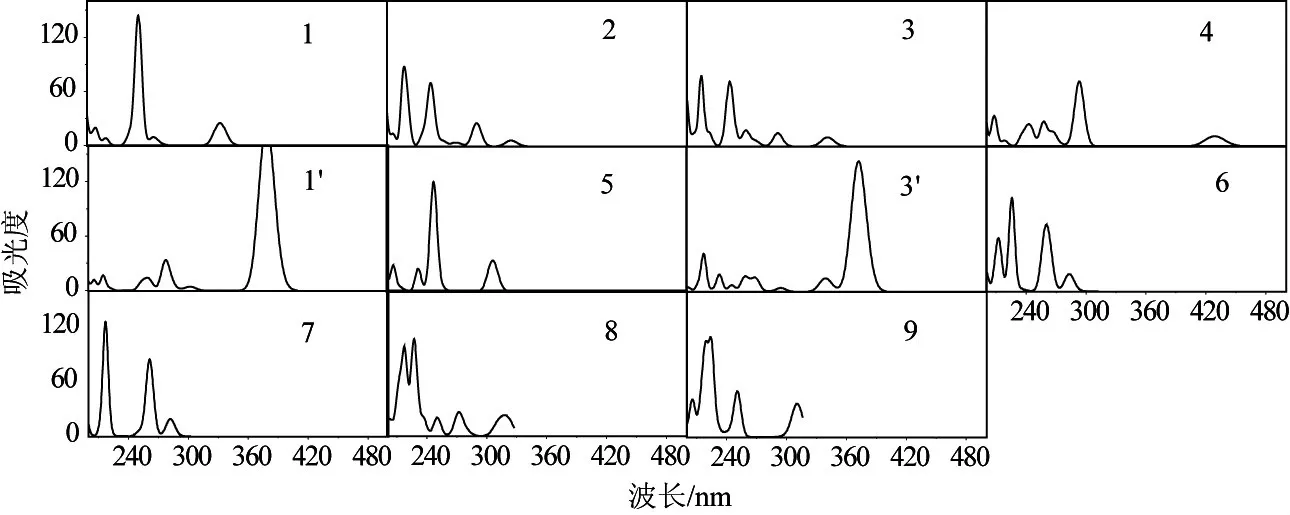
图2 各分子的电子吸收光谱
偶极矩表征分子内电子分布的不均匀性和不对称性,极化率表征电子分布在外场下的变化,均为表征分子电子性质的重要参数。表2列出了计算得到的各个分子的基态偶极矩和极化率,除3以外的其余4个分子均具有较大的偶极矩,这是由于这些分子的取代基的供/吸电子能力及位置不同造成的。在这些分子中,供电子的氨基和甲氧基处于Si-Si键的对位。1,4,5中吸电子的氰基和硝基也处于Si-Si的对位,形成较大的电荷分离,因此具有较大的偶极矩。2中氰基居间位,偶极矩次之,3的氰基居邻位,偶极矩最小。这些分子的极化率相差不大,其中3的极化率最小。值得注意的是,它们的极化率和1'和3'的极化率无明显差异,反映出共轭体系被Si-Si阻碍并没有严重影响电子在2个苯环之间的转移。
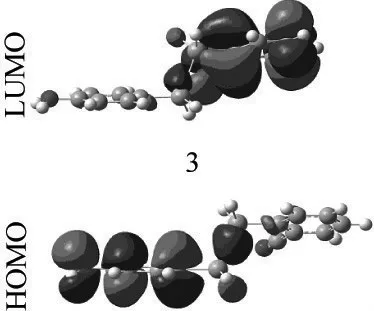
图3 结构3的HOMO和LUMO轨道
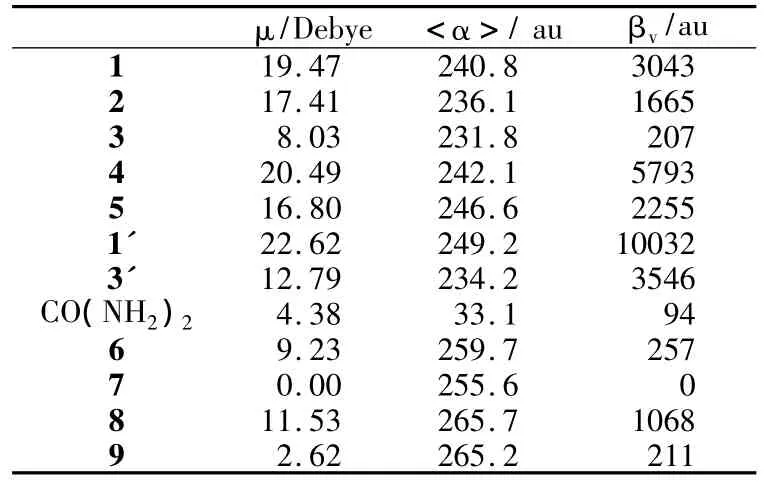
表2 分子的偶极矩(μ)、极化率(<α>)和超极化率(βv)
表2中的超极化率的计算公式为:
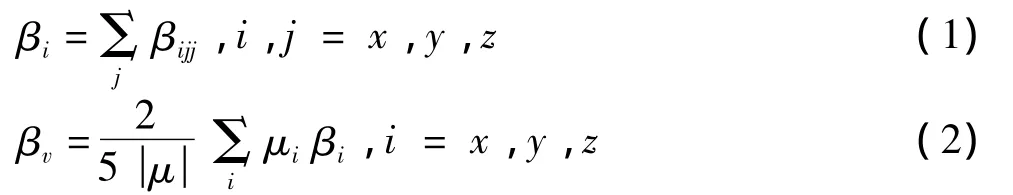
其中:βijj为利用有限场方法计算得到的超极化率张量元;βv是沿偶极矩方向投影的超极化率数值,对应于二次谐波产生(SHG)过程。除3以外,其余各分子具有很大的响应系数,这反映出电子在激发态下可以较容易地在供电子和吸电子基团之间转移,具有较小的激发能和较大的跃迁几率。其原因是Si中的3d轨道和苯环中碳原子的2p轨道可以发生重叠,饱和的Si-Si键并没有完全阻断2苯环之间的电子传递。3的超极化率数值较小,是因为它在间位的氰基吸引了电子,影响了电子在苯环中的大范围移动。虽然3的超极化率小于其他分子,但其数值仍为尿素分子的2倍多(实验表征值为2.9倍[6]),表明3是一个很有应用价值的有机NLO材料。
有意思的是,在1~5中,仅有3被表征出NLO活性。其余分子虽然具有很大的超极化率数值,却没有NLO活性。这源于它们具有较大的偶极矩,在形成晶体时,这些有机分子受到较强的静电及偶极作用,被迫交替反向排列,形成了具有对称中心的晶体结构,失去了二阶NLO活性。作为对比的分子1'和3'的情况类似,也没有NLO活性。3具有较小的偶极矩,分子间偶极-偶极相互作用较弱,在结晶过程中可以旋转的Si-Si键可以帮助它避免形成对称中心。因此,3的晶体保留了NLO活性。
按照上述原理,我们在1~5的基础上设计了4个参考分子6~9,它们保留了Si-Si键,但在两侧苯环中分别引入了供电子和吸电子基团,在Si-Si的两侧分别形成小的推拉型子结构。其设计原理是:1)引入取代基减小偶极矩;2)保持体系的共轭性;3)保留Si-Si键,以保持透光性和结晶过程中分子的可旋转性。计算得到的6~9的电子吸收光谱见图2,偶极矩、极化率和超极化率见表2。4个分子的第一吸收波长均小于320 nm,具有良好的透光性。7由于具有对称中心,其超极化率为0。6,8和9具有相对较小的偶极矩,具有与3相近或者更大的超极化率。因此,这3种设计分子具有较大的NLO响应,在近紫外及可见光区具有良好透光性,且满足非线性光学晶体结晶过程的要求,是一类值得进一步进行实验研究的NLO备选材料。
3 结语
用密度泛函理论的B3LYP泛函和6-311++G(d,p)基组计算研究了5个内嵌乙硅烷的推拉型有机分子的结构特征、电子吸收光谱和NLO性质。含较大共轭体系的推拉型有机分子往往具有较大的偶极矩、极化率和超极化率,但透光性较差。由于在共轭桥中的乙硅烷含有饱和的Si-Si键,改变了基态和激发态分子体系内的电子分布状态,本文研究的5种化合物分子都具有较好的透光性和NLO响应。分析发现,由于Si原子的3d轨道对共轭体系有贡献,饱和的Si-Si键只是部分地影响了其两端苯环之间的电子转移,导致了电子第一吸收峰蓝移。同时,Si-Si单键具有较好的空间旋转能力,在结晶过程中有助于避免形成含有对称中心的晶体结构。根据上述原理,我们设计了3个具有较低偶极矩、较好透光性、较大超极化率的含乙硅烷的推拉型有机分子,供实验科学家制备和验证其NLO效应。
[1]GESKIN V M,LAMBERT C,BREDAS J L.Origin of high second-and third-order nonlinear optical response in Ammonio/Borato diphenylpolyene zwitterions:the remarkable role of polarized aromatic groups[J].J.Am.Chem.Soc.,2003,125(50):15651-15658.
[2]ANDREU R,BLESA M J,CARRASQUER L,et al.Tuning first molecular hyperpolarizabilities through the use of proaromatic spacers[J].J.Am.Chem.Soc.,2005,127(24):8835-8845.
[3]HUMPHREY J L,LOTT K M,WREGHT M E,et al.Second hyperpolarizability of ethynyl-linked Azobenzene molecular wires[J].J.Phys.Chem.B,2005,109(46):21496-21498.
[4]LIU C G,QIU Y Q,SU Z M,et al.Computational study on secondorder nonlinear response of a series of two-dimensional carbazole-cored chromophores[J].J.Phys.Chem.C,2008,112(17):7021-7028.
[5]ALBERT I D L,MARKS T J,RATNER M A.Conformationally-Induced geometric electron localization.interrupted conjugation,very large hyperpolarizabilities,and sizable infrared absorption in simple twisted molecular chromophores [J].J.Am.Chem.Soc.,1997,119(13):3155-3156.
[6]SHIMADA M,YAMANOI Y,MATSUSHITA T,et al.Optical properties of Disilane-Bridged Donor-Acceptor Architectures:strong effect of substituents on fluorescence and nonlinear optical properties[J].J.Am.Chem.Soc.,2015,137:1024-1027.
[7]LEE C,YANG W,PARR R G.Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density[J].Phys.Rev.B,1988,37(2):785-789.
[8]BAUERNSCHMITT R,AHLRICHS R.Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory[J].Chem.Phys.Lett.,1996,256:454-464.
[9]JAMORSKI C,CASIDA M E,SALAHUB D R.Dynamic polarizabilities and excitation spectra from a molecular implementation of timedependent density-functional response theory:N2 as a case study[J].J.Chem.Phys.,1996,104(13):5134-5147.
[10]CEHEN H D,ROOTHAAN C C J.Electric dipole polarizability of atoms by the Hartree-Fock method.I.theory for Closed-Shell systems[J].J.Chem.Phys.,1965,43(10):34-39.
[11]KURTZ H A,STEWART J J P,DIETER K M.Calculation of the nonlinear optical properties of molecules[J].J.Comput.Chem,1990,11:82-87.
[12]FRISCH M J,TRUCKS G W,SCHLEGEL H B,Gaussian 09,Revision A.1[Z].Gaussian,Inc.,Wallingford CT,2009.
猜你喜欢
纺织报告(2022年10期)2022-11-04
中学化学(2022年5期)2022-06-17
光子学报(2022年3期)2022-04-01
贵州大学学报(自然科学版)(2022年1期)2022-01-26
口腔医学(2020年8期)2020-09-08
高中数理化(2020年1期)2020-02-29
科学之谜(2019年9期)2019-10-16
理科考试研究·高中(2019年8期)2019-09-19
价值工程(2017年6期)2017-03-15
化学教学(2015年11期)2015-12-19
