
利用分子理论估算四氢呋喃的热物理特性参数
2015-03-14 03:22樊祥山孙德魁王井山王锡斌
西安交通大学学报 2015年3期
樊祥山,孙德魁,王井山,王锡斌
(西安交通大学能源与动力工程学院,710049,西安)
利用分子理论估算四氢呋喃的热物理特性参数
樊祥山,孙德魁,王井山,王锡斌
(西安交通大学能源与动力工程学院,710049,西安)
为了开展四氢呋喃作为发动机代用燃料的喷雾燃烧的数值模拟,对四氢呋喃的密度、黏度、表面张力、蒸气压、气化潜热、比热容、导热系数、扩散系数等随温度变化的热物理特性参数进行了估算,并基于分子理论提出了四氢呋喃各参数与温度的关系式。估算过程中应用了对比态原理与基团贡献法,通过分子状态的相对特性值或分子结构预测出了相应物性,对分子偏心带来的影响进行了修正。通过与实验值的比对,各参数关系式的最大误差均小于10%,可满足喷雾和燃烧过程数值模拟的要求;四氢呋喃在能量密度、黏度、饱和蒸气压和沸点等方面具有的优势,使其有望成为内燃机的代用燃料;运用分子理论,在较少实验值的基础上可以得到满足数值模拟要求的热物性数据。
四氢呋喃;热特性;分子理论
化石燃料消耗引起的能源枯竭以及环境恶化使生物质燃料受到越来越多的重视,第二代生物质燃料不再以粮食为原料,转而使用非粮作物。四氢呋喃(tetrahydrofuran,THF)是一种有机合成原料,是性能优良的溶剂,被广泛应用于有机化学工业和制药工业,目前主要的工业生产方法有糠醛法、雷由法(Reppe法)和以顺酐为原料的催化加氢法等[1],炼制原料包括秸秆、玉米芯、木屑等生物质,生产成本较低,符合第二代生物质燃料特征。四氢呋喃分子中含有氧元素(C4H8O),无不饱和碳键,其与乙醇相比在能量密度、黏度、气化潜热、沸点等特性上有较大优势,可以作为内燃机生物质燃料和助溶剂使用。为了探究这种物质在内燃机中应用的潜力,有必要对其相关特性进行理论与实验研究。
燃料的喷雾和燃烧性能会影响内燃机的工作方式和工作特性,所以探究四氢呋喃的喷雾和燃烧特性,即在大测试范围内针对新型燃料进行喷雾和燃烧的数值模拟及台架实验具有重要意义。
数值模拟时需要预知燃料的热物性参数,然而在内燃机工作过程中燃料经历了大跨度的温度及压力变化,加热是由燃料喷入气缸到燃烧中后期的过程,若采用数值方法模拟如此大的工况范围,各种物性参数必须作为变量来考虑。对于液态燃料,需要定量地预知其在常温到临界温度之间的各种热物性参数以及转变为气相之后的某些重要热物性参数。
文献[2-3]成功地基于分子理论对二甲醚和生物柴油的热物性进行了大温度范围的预测。目前,四氢呋喃热物性的实验测定工作很少,文献[4-6]涉及到了参考值,文献[7-10]对液相四氢呋喃在某些温度下的黏度和密度进行了测量。本文利用分子理论对四氢呋喃的热物性进行了预测,为后续的数值模拟做准备。
1 基本物理性质
通过查阅文献,得到了四氢呋喃在某些特定条件下的基本参数,并在表1中与其他常用燃料进行了对比。

表1 四氢呋喃与多种燃料的特性参数对比
注:着火极限为空气中可燃气体积比。
2 采用分子理论估算四氢呋喃的热物性参数
对大跨度温度变化的内燃机工作过程进行气缸内(尤其是喷雾过程)的多维数值模拟,需要掌握不同温度下液体燃料的热物性,包括密度、沸点、黏度、蒸气压、气化潜热、表面张力、焓、导热系数、扩散系数等,这些参数均随温度、压力等状态参数的变化发生很大变化,仅靠表1数据远不能满足需求,所以本文通过热力学理论或根据实验值进行预测。估测值并非精确,但在特性参数缺乏而客观上又必需要求时可以满足或部分满足数值模拟的需要。
2.1 分子理论
预测热物性的过程中用到的经验公式是通过观察物质的共性提出的,公式中的经验参数根据不同物质将发生相应的变化来体现各种物质宏观上的差异性,这些差异性通常归结于物质微观结构的差异。
分子理论已大量应用到未知流体热物性的估测中,该理论认为流体的宏观特性与微观分子的结构、大小及运动速度等密切相关。采用分子理论估算流体热物性时涉及到了对比态原理、分子结构、分子极性等[2]。
运用对比态原理对四氢呋喃未知热物性进行估测时涉及到的相对特性值包括相对温度Tr=T/Tc和相对沸点温度Tbr=Tb/Tc,其中Tb与Tc分别是四氢呋喃的沸点温度和临界温度。另外,运用基团贡献法时需要了解四氢呋喃(C4H8O)的含氧五元杂环的分子结构,该分子结构中有一个非烃戊环基团,其中包括1个醚基—O—基团和4个—CH2—基团,属于环醚类化合物。通过综合考虑,本文估算出了四氢呋喃的各种经验参数,由此提出了四氢呋喃各种热物性的计算公式,从四氢呋喃中等极性(偏心因子0.226[14])看,计算效果良好。
2.2 液态导热系数
采用Sastri方法[5]、结合对比态原理和基团贡献法计算了液相四氢呋喃的导热系数
λL=λbam
(1)
(2)
式中:λb为标准沸点导热系数;a和n均为拟合参数。对于环醚类化合物,a=0.16,n=0.2。λb可以由基团贡献和修正值求得,见表2。
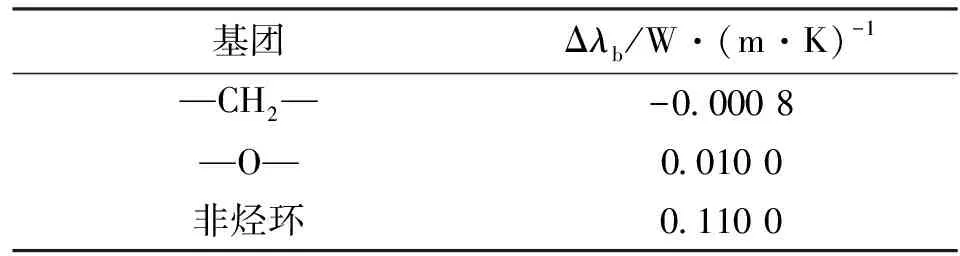
表2 采用Sastri方法获得的基团贡献值
由于四氢呋喃有4个—CH2—基团、1个—O—基团、1个非烃环,因此
λb=4×(-0.000 8)+0.010 0+0.110 0=
0.116 8 (W/(m·K))
于是,最终的四氢呋喃的液态导热系数计算式为
(3)
由于缺乏四氢呋喃液态导热系数的实验参考值,所以在此无法估算液态导热系数的计算误差。但是,根据文献[5]中对多种物质的误差分析发现,计算结果的平均误差为8%,尚在可以接受的范围之内。
2.3 表面张力
采用对比态理论对表面张力(mN/m)进行了计算[6],即
σ=A(1-T/Tc)N
(4)
式中:A和N均为拟合参数。通过大量的数据收集和回归统计得到的四氢呋喃的拟合参数A=71.103、N=1.222 2,Tc=540.15 K[6],所以最终的表面张力计算式为
σ=71.103×(1-Tr)1.222 2
(5)
根据文献[5,13],四氢呋喃在303 K和298 K时的表面张力分别为26.4 mN/m和28 mN/m,本文模拟值分别采用26.00 mN/m和26.67 mN/m,二者偏差分别是1.52%和4.75%。
2.4 黏度计算
温度低于或高于380 K(Tr≈0.7),四氢呋喃黏度应用了2个计算公式。低温时对黏度实验数据进行了拟合,拟合公式采用Andrade方程[15],即
(6)
拟合数据见表3。拟合后可求出如下近似值
C=-4.498 5
B=1 110.7
于是得到低温时动力黏度(mPa·s)
(7)
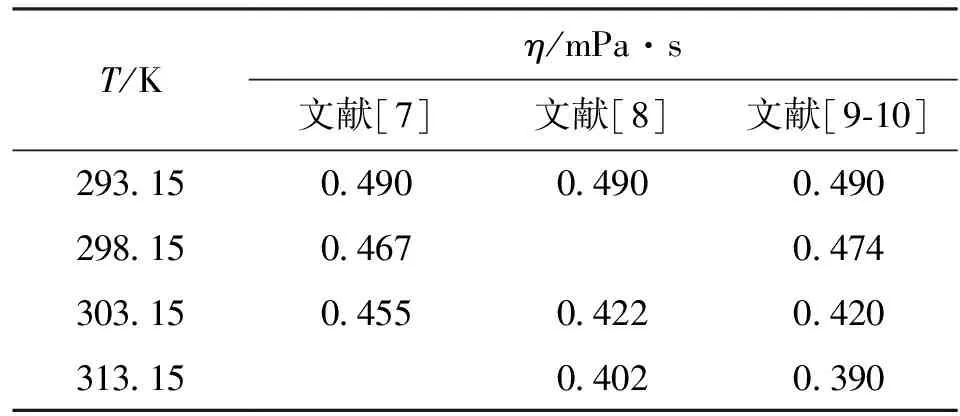
表3 四氢呋喃低温动力黏度拟合数据
经对比发现,本文计算结果与文献[7-10]中实验数据吻合较好。如图1所示,低温阶段动力黏度的计算值与实验值的最大误差为4.61%,在允许的范围内。
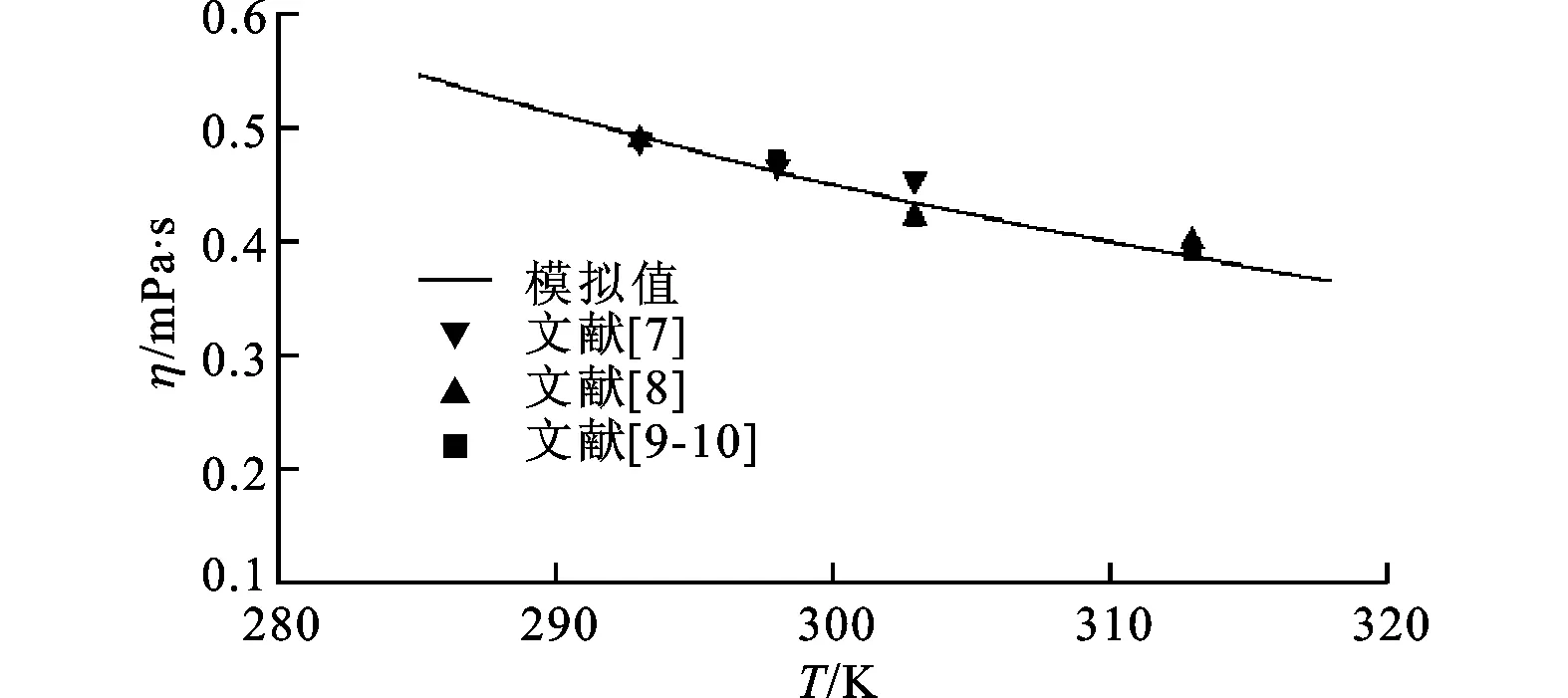
图1 四氢呋喃低温黏度模拟值和实验值对比
高温时动力黏度采用了Sastri方法的基于对比态原理的计算公式[5],即
(8)
(9)
对于四氢呋喃,Tbr=Tb/Tc=0.628;对于醇类,式(8)中α=0.1175,对于其他化合物α=0.248。ηb是沸点下的黏度,可以按表4由基团贡献法求得。
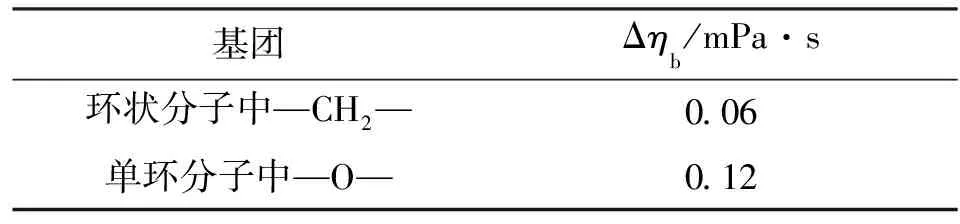
表4 Sastri方法计算ηb的基团贡献值
四氢呋喃是单环分子,含有1个醚基—O—基团和4个—CH2—基团,属于环醚类有机物,其α=0.248,ηb=0.06×4+0.12=0.36 (mPa·s)。
高温下四氢呋喃动力黏度的最终拟合公式为
lnη=-2.46×0.415Φ
(10)
(11)
Sastri报告中指出[5],对于Tr>0.9,动力黏度计算值与实验值的平均偏差为10%,对于Tbr 2.5 密度计算 液相四氢呋喃密度计算采用了基于对比态原理的公式[6],即 ρ=UV-(1-T/Tc)γ (12) 式中:U、V和γ都是经验参数。文献[6]得到了四氢呋喃的U=0.322,V=0.280 78,γ=0.291 2。由此,密度的最终计算公式为 ρ=0.322×0.280 78-(1-Tr)0.291 2 (13) 从文献[7-8,10]中可以整理出两组实验数据,经过与模拟得到的数据比对,发现二者非常吻合且最大误差为0.48%,在容许的范围之内,如图2所示。 图2 四氢呋喃密度模拟值和实验值对比 2.6 饱和蒸气压 四氢呋喃饱和蒸气压(Pa)的计算采用了Antoine[16]方程,即 lgPvap=E-F/(T+G) (14) 式中:E、F和G是拟合参数。由参考文献[5]可知,对于四氢呋喃,式(14)的适用温度为253.5~361.71K,E=9.121 42、F=1 203.110、G=-46.795,由此得到 lgPvap=9.121 42-1 203.11/(T-46.795) (15) 由式(15)获得的饱和蒸气压与文献[5]结果的误差为1.59%,在容许的范围之内。 2.7 气化潜热 气化潜热采用了文献[6]的基于对比态原理的计算公式,即 ΔHvap=p(1-Tb/Tc)q (16) 对于四氢呋喃,p=44.439,q=0.391[6]。所以,四氢呋喃的气化潜热(kJ/mol)为 ΔHvap=44.43×(1-Tbr)0.391 (17) 根据文献[6],四氢呋喃在常压下的338K时,气化潜热实验值为30.260kJ/mol,计算值为30.20kJ/mol,二者误差为0.20%,满足要求。 2.8 比焓计算 物质比焓的计算式为 (18) (19) (20) 式中:R为气体常数。 根据文献[5],式(20)中的拟合系数a0=5.171×10-3,a1=-19.464×10-6,a2=16.46×10-8,a3=-20.42×10-11,a4=8×10-14,适用范围为50~1 000K。对于1 000K以上的高温,由于数据有限以及燃料的高温分解等原因,所以暂时不能模拟,还需要通过数据搜集和实验来获得。 四氢呋喃比焓(kJ/mol)最终相关计算式为 (T-298.15 K),T (21) (Tb-298.15)+44.439 kJ/mol× (22) (23) 16.46×10-8T2-20.42×10-11T3+8×10-14T4) (24) 通过比对文献[5]中的比热容数据,发现计算误差为1.64%,在可接受的范围之内。 2.9 扩散系数 气相四氢呋喃在空气中的扩散系数可根据文献[15]公式计算,即 (25) 式中:M1和M2与(Σv)1和(Σv)2分别为2种组分的相对分子质量与分子扩散容积。按表5数据可以得到四氢呋喃的分子扩散容积 (Σv)THF=4×15.9+8×2.31+6.11-18.3= 69.89 (cm3/mol) 对于空气,(Σv)air=19.7cm3/mol,MTHF=72.107,Mair=28.96。 表5 原子扩散容积的基团贡献值 经过计算可得四氢呋喃在空气中的扩散系数(cm2/s)为 (26) 由于缺乏四氢呋喃在空气中的扩散系数实验参考值,故计算误差还无法进行估算。文献[5]指出,式(26)的平均绝对误差为4%,这也在可以接受的范围之内。 通过对四氢呋喃物理性的计算和分析,可以得到以下结论。 (1)四氢呋喃的生产过程和物性基本满足替代燃料的标准,其能量密度、黏度、饱和蒸气压和沸点温度等具有优势,有望成为内燃机的代用燃料。 (2)内燃机工作过程中,燃油的各种物性参数变化很大,必须按变量来处理,而四氢呋喃的宏观热物性可以利用分子理论、根据分子结构特性估算出来。 (3)本文应用分子理论估算出了四氢呋喃的密度、表面张力、潜热、蒸气压、导热系数、扩散系数等随温度的变化,并给出了最终计算关系式。通过对比部分实验值,观察到关系式的最大误差小于10%,满足内燃机工作过程的多维模拟的要求。 [1] 张希功. 四氢呋喃技术进展与生产现状 [J]. 化工生产与技术, 2002, 9(2): 18-20. ZHANG Xigong. Production technology progress and status of tetrahydrofuran [J]. Chemical Production and Technology, 2002, 9(2): 18-20. [2] 王锡斌, 蒋德明, 周龙保, 等. 利用分子理论估算二甲醚的热物理特性参数 [J]. 内燃机学报, 2004, 22(6): 486-492. WANG Xibin, JIANG Deming, ZHOU Longbao, et al. Estimation of dimethyl ether thermal properties by molecule theory [J]. Transaction of CSICE, 2004, 22(6): 486-492. [3] 阮登芳, 王文彬, 吴长河. 生物柴油物理特性参数的估算与误差分析 [J]. 大豆科学, 2009, 28(6): 1072-1075. RUAN Dengfang, WANG Wenbin, WU Changhe. Physical property prediction of bio-diesel and error analysis [J]. Soybean Science, 2009, 28(6): 1072-1075. [4] SMALLWOOD I M. Handbook of organic solvent properties [M]. New York, USA: Halsted Press, 1996: 217. [5] POLING B E, PRAUSNITZ J M, CONNELL J P O. The properties of gases and liquids [M]. 5th ed. New York: McGraw-Hill, 2001: 9. [6] YAWS C L. Thermophysical properties of chemicals and hydrocarbons [M]. New York, USA: William Andrew, 2008: 12-696. [7] KINART C M, KINART W J, EWIKLINSKA A. 2-methoxyethanol tetrahydrofuran binary liquid system viscosities, densities, excess molar volumes and excess Gibbs activation energies of viscous flow at various temperatures [J]. Journal of Thermal Analysis and Calorimetry, 2002, 68: 307-317. [8] PARVEEN S, SHUKLA D, SINGH S, et al. Ultrasonic velocity, density, viscosity and their excess parameters of the binary mixtures of tetrahydrofuran with methanol and o-cresol at varying temperatures [J]. Applied Acoustics, 2009, 70(3): 507-513. [9] GUPTA M, VIBHU I, SHUKLA J P. Ultrasonic velocity, viscosity and excess properties of binary mixture of tetrahydrofuran with 1-propanol and 2-propanol [J]. Fluid Phase Equilibria, 2006, 244(1): 26-32. [10]DUBEY G P, DENSITIES R K. Densities, speeds of sound and viscosities of binary mixtures of tetrahydrofuran with 1-hexanol, 1-octanol and 1-decanol atT=(298.15 to 313.15) K [J]. Chem Thermodynamics, 2014, 71: 27-36. [11]蒋德明, 黄佐华. 内燃机替代燃料燃烧学 [M]. 西安: 西安交通大学出版社, 2007: 4-27. [12]田国弘, 徐宏明, DANIEL R , et al. 2, 5-二甲基呋喃的喷雾特性及发动机适应性 [J]. 汽车安全与节能学报, 2010, 1(2): 132-140. TIAN Guohong, XU Hongming, DANIEL R, et al. Spray characteristics and engine adaptability of 2, 5-dimethylfuran [J]. Journal of Automotive Safety and Energy, 2010, 1(2): 132-140. [13]程能林. 溶剂手册 [M]. 4版. 北京: 化学工业出版社, 2007: 559-561. [14]卢焕章. 石油化工基础数据手册 [M]. 北京: 化工出版社, 1984: 946. [15]童景山. 流体热物性学 [M]. 北京: 中国石化出版社, 2008: 267-330. [16]陈钟秀, 顾飞燕, 胡望明. 化工热力学 [M]. 3版. 北京: 化学工业出版社, 2012: 289. [17]张宇英, 张克武. 分子热力学性质手册 [M]. 北京: 化学工业出版社, 2009: 362. (编辑 苗凌) Estimation for Thermal Properties of Tetrahydrofuran with Molecule Theory FAN Xiangshan,SUN Dekui,WANG Jingshan,WANG Xibin (School of Energy and Power Engineering, Xi’an Jiaotong University, Xi’an 710049, China) To numerically simulate the spray and combustion process of tetrahydrofuran (THF) as an alternative fuel of internal combustion engine, the correlations of thermo-physical properties versus temperature, including density, viscosity, surface tension, vapor pressure, latent heat value, heat capacity, thermal conductivity and diffusion coefficient, are estimated with molecule theory. The estimations are conducted with group-contribution method following the corresponding state theory and the effect from polar molecule structure is modified. The comparison with experimental data indicates that the maximum deviations of all the estimated correlations are much less than 10%, and the estimation thus meets the requirements of numerical simulation on THF spray and combustion process. THF is a promising alternative fuel for internal combustion engine due to the advantages in energy density, viscosity, vapor pressure, and the boiling point. tetrahydrofuran; thermal properties; molecule theory 2014-06-25。 作者简介:樊祥山(1992—),男,硕士生;王锡斌(通信作者),男,副教授。 基金项目:国家自然科学基金资助项目(61235003)。 时间: 2015-01-05 网络出版地址: http:∥www.cnki.net/kcms/detail/61.1069.T.20150105.0855.006.html 10.7652/xjtuxb201503008 TK407.9 A 0253-987X(2015)03-0044-06
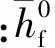
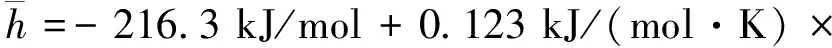

3 结 论
猜你喜欢
化工设计(2022年4期)2023-01-02贵州科学(2022年4期)2022-09-05材料与冶金学报(2022年2期)2022-08-10能源工程(2022年1期)2022-03-29韩国语教学与研究(2021年1期)2021-07-29煤气与热力(2021年6期)2021-07-28新课程·下旬(2019年7期)2019-09-17现代盐化工(2019年4期)2019-09-10铜仁学院学报(2018年6期)2018-07-05浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
