
单环环烷烃分子水平催化裂化反应动力学模型的建立
2015-07-02 01:39王鑫磊郭锦标
石油学报(石油加工) 2015年6期
张 旭, 周 祥, 王鑫磊, 郭锦标
(中国石化 石油化工科学研究院, 北京 100083)
单环环烷烃分子水平催化裂化反应动力学模型的建立
张 旭, 周 祥, 王鑫磊, 郭锦标
(中国石化 石油化工科学研究院, 北京 100083)
在深入研究环烷烃催化裂化反应机理的基础上,采用反应族的概念制定了11大类、37条针对单环环烷烃催化裂化反应的规则,并结合计算化学建立了基于反应路径层面的单环环烷烃分子水平催化裂化反应动力学模型。以辛基环己烷为模型化合物,验证了建立的动力学模型,任选4组实验数据回归得到了16类催化裂化反应动力学参数值。结果表明,该模型能预测环烷烃催化裂化的主要反应产物分布,预测值与实验值吻合良好;得到了4种反应类型在不同空速下对转化率的贡献值。另外,回归得到的动力学参数值可以用于其他单环环烷烃催化裂化反应体系,为进一步建立其他环烷烃催化裂化反应动力学模型提供数据支持。
分子水平; 动力学模型; 动力学参数; 单环环烷烃; 催化裂化
当前,日益严格的环保法规一方面要求减少污染物排放总量,另一方面还明确了车用燃油产品的各项具体质量指标。如国Ⅴ汽油和柴油质量标准规定,硫质量分数不超过10 μg/g,对苯、整体芳烃的含量也设定了上限值[1]。另外,随着原油开采的不断推进,原油重质化、劣质化的趋势也愈发明显。为了尽可能多地将重质、劣质原油转化为轻质油品,采用加氢-催化裂化联合工艺生产清洁汽、柴油。这一加工过程是将稠环芳烃变成环烷烃,再到小分子烷烃、烯烃及芳烃的过程。为了更好地利用石油资源,指导新工艺开发,需要将对石油及其转化规律的认识从传统的集总水平上升到分子水平,也要求动力学模型的发展上升到分子水平。分子水平动力学模型能够对复杂反应系统中的产物分布及其性质的预测更准确,并且能反映出原料性质、催化剂、操作变量三者之间的内在联系[2]。
研究催化裂化反应动力学模型对认识催化裂化反应过程、工艺开发和过程控制都起到至关重要的作用。模型经历了关联模型[3]、集总动力学模型[4-7]到分子水平动力学模型[8-10]3个发展阶段。关联模型在分析水平较低的时候得到了广泛的应用。集总动力学模型用数量有限的虚拟组分来描述反应过程的化学计量学变化,现在仍是认识催化裂化反应过程和指导催化裂化工艺开发的主要方法;由于没有赋予虚拟组分化学意义,所以预测能力有限,外推性较差,最大的不足在于模型不能够提供详细的石油分子组成及结构信息,不能满足催化裂化新工艺开发需求。过去二十年,由于分析水平、计算机数据处理能力的不断提高,促进了分子水平动力学模型的迅速发展。另外,分子水平动力学模型不仅能够帮助人们进一步认识反应机理,还能定量预测更多的分子细节。
20世纪90年代就开始研究烷烃[11-12]、烷基苯[13]等模型化合物的分子水平动力学,它的发展也面临着诸多的挑战。比如,如何定量描述复杂反应体系中各个反应的反应速率、吸附速率、平衡常数等。烃类化合物的催化裂化反应数量成百上千,反应过程中还存在着耦合反应,构成的反应网络极其复杂。面对数量庞大的原料和产物体系,如何建立一个形式简单,又能够提供足够多反应信息的分子水平动力学模型十分困难。先后出现了结构化模型(Structural model,SM)、结构导向型集总模型(Structure oriented lumping,SOL)[10]、单事件模型(Single event kinetic model,SEKM)[8]、机理模型[11-13]等。SM模型适合于烷烃体系,对于含有环烷环、芳环结构分子的预测效果较差;SEKM模型需要较为深厚的理论基础,计算量大,难以实际操作;SOL模型需要有大量的分析数据,且理论基础较薄弱。所以,需要开发一种形式简单、计算量小、能够提供更多细节信息的分子水平催化裂化动力学模型。
环烷烃可以脱氢生成芳烃,也可以开环生成小分子的烷烃、烯烃以及烷基环烷烃。然而,以往大多侧重馏分油的集总水平催化裂化动力学模型,不能清晰地反映各组分之间的变化规律。为此,笔者在环烷烃分子水平动力学模型前期研究的基础上,以辛基环己烷为模型化合物,探索建立基于反应路径层面单环环烷烃催化裂化分子水平反应动力学模型。
1 实验部分
1.1 原料和催化剂
辛基环己烷(OCHA),质量分数99%以上,Aldrich公司产品。
实验用催化剂由质量分数为40%REY分子筛、20%硅溶胶以及40%高岭土组成,经喷雾干燥、成型、焙烧、老化处理等工序制备得到。REY分子筛中Si/Al摩尔比为2.7,比表面积103 m2/g、孔容0.11 cm3/g。老化后催化剂经XRD测得其晶胞大小为2.44 nm。
1.2 实验及数据处理方法
采用等温固定床反应器评价催化剂活性[14]。在建立分子水平动力学模型时,以摩尔流率(mol/s)来表示反应体系中各组分的浓度,因此,需要将质量浓度换算为摩尔浓度[15]。
2 建立分子水平催化裂化反应动力学模型基本步骤
分子水平动力学模型主要包括4个组成部分[15]。
首先是构建原料分子组成模型。对于较简单原料而言,通过普通的表征就可以获得原料的分子组成。对于复杂的反应原料,通常需要核磁共振(NMR)、气相色谱-质谱联用(GC-MS)等更加详细的分析手段。
第2部分是建立反应模型。通过分子管理和制定反应规则,结合反应族的概念,建立数量庞大的分子反应网络。
第3部分是动力学参数(反应速率常数)的求取。动力学参数主要是阿伦尼乌斯方程的指前因子、活化能。可以通过张旭等[16]和杜梅西克等[17]建立的方法获得指前因子,由量子化学计算软件—Material Studio(MS)软件DMol3模块计算则可获得活化能。
最后一部分是模型的求解和应用。采用Levenberg-Marquardt(LM)算法回归计算,通过不断调整参数,使得模拟值与实验值的方差逐渐缩小,当达到设定收敛精度,则停止计算,否则继续调整动力学参数值,重复计算直到满足设定收敛精度为止。当获得产物分布后,经过计算可获得产物的宏观性质数据。
3 单环环烷烃分子水平催化裂化反应动力学模型的建立
3.1 反应数据处理
表1列出了6种不同空速下,OCHA在REY催化下催化裂化反应的转化率、生焦量等数据。表2列出了主要反应产物的摩尔流率。由于分析手段的限制,不能对每一产物的分子结构鉴别,需进行一定的归并处理。如将取代烷烃都看作是二位单甲基取代;所有烯烃看作是一位烯烃;C8环烷烃看作是乙基环己烷(ECHA),C9环烷烃统一归为丙基环己烷(PCHA),C10环烷烃记为正丁基环己烷(BCHA);含苯环的C8记为乙苯(EBEN),含苯环的C9记为丙基苯(PPBEN),含苯环的C10记为丁基苯(BBEN),含苯环的C11记为正戊基苯(PEBEN)。
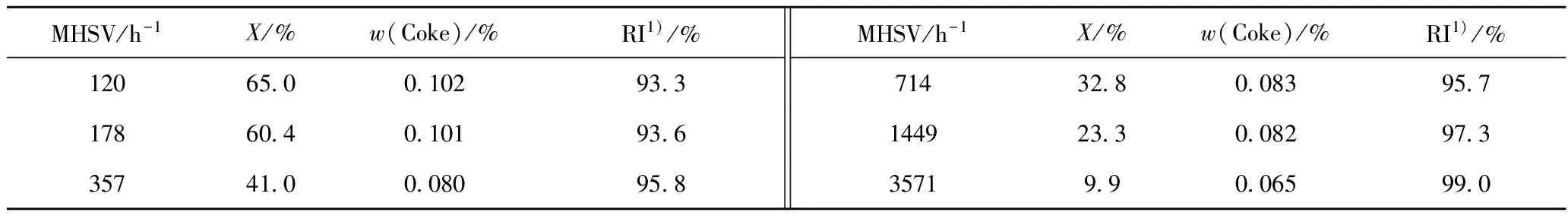
表1 不同空速(MHSV)下REY催化OCHA催化裂化反应性能
1) Product recovery index
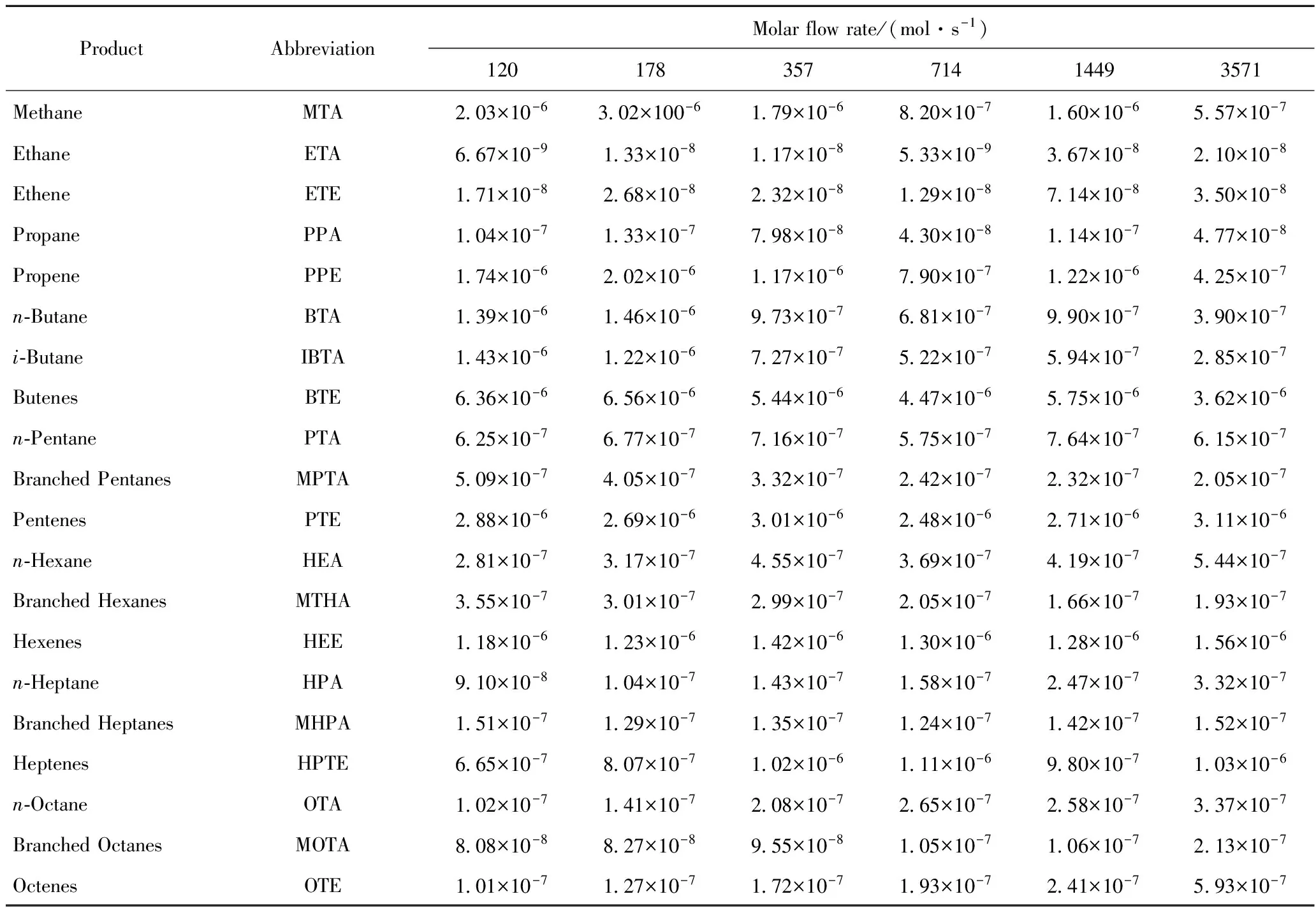
表2 不同空速下REY催化OCHA催化裂化反应产物摩尔流率[14]
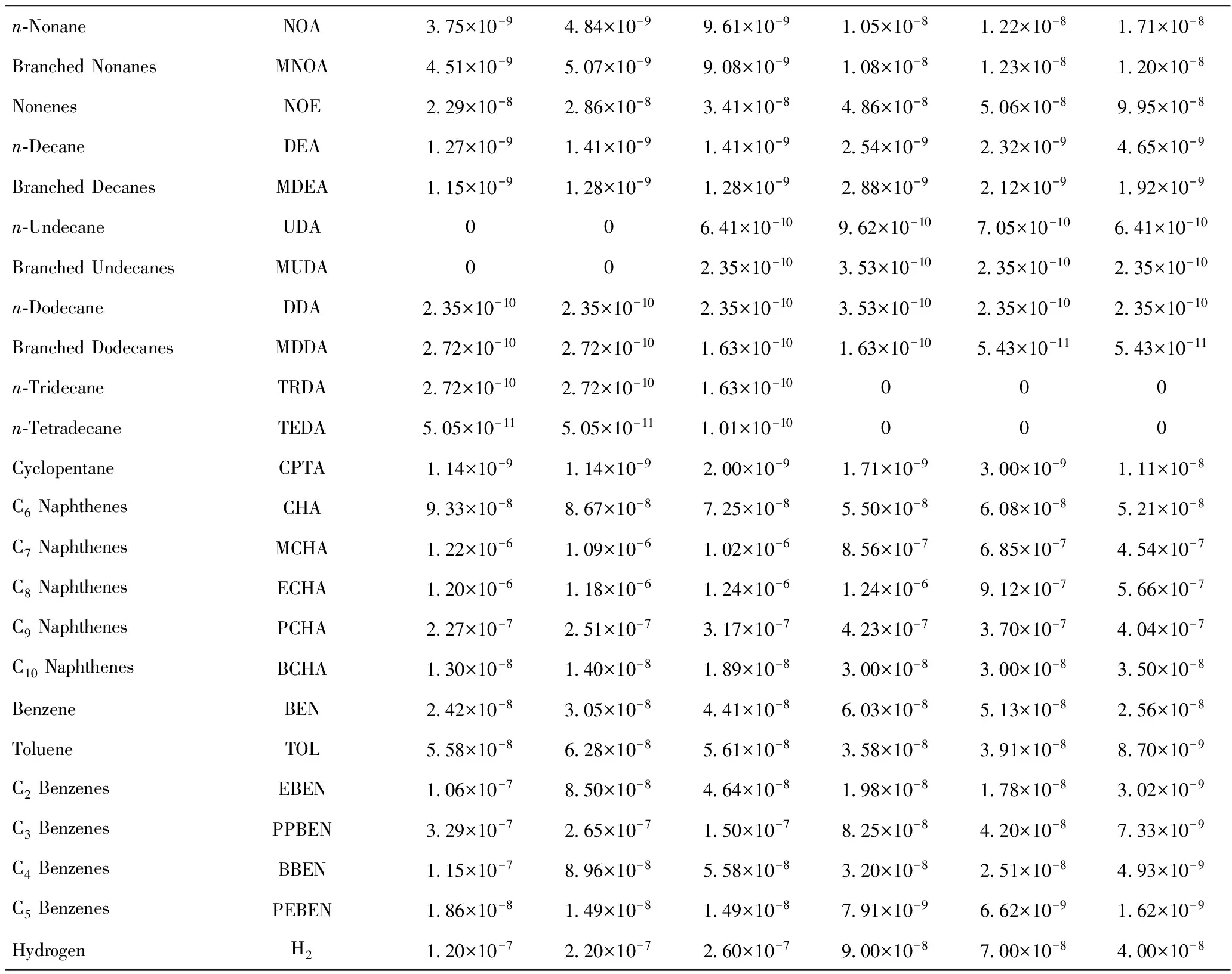
续表2
3.2 反应路径分析
OCHA烷基侧链裂化可以产生烷烃、烯烃以及C6~C10环烷烃,开环后会发生二次裂化,产生C3~C8烷烃和烯烃。即使在低转化率情况下,OCHA的二次裂化反应速率仍很快,因为在产物中没有检测到 C14产物。随着分子中碳原子数增加,烷烃在热力学上容易发生裂化反应。
含7个及以上碳原子的正构烷烃和含8个及以上碳原子的异构烷烃可以发生二次裂化反应,产生含更少碳原子的分子。含有4个及以上碳原子的烯烃也经历了二次裂化反应,如氢转移或裂化反应。含有9个或9个以上碳原子的环烷烃可以进一步发生反应,形成短链的环烷烃或脱氢生成芳烃,芳烃和烯烃可能是焦炭的前躯体。
3.3 反应规则的制定
反应规则包含2个方面的内容,一个是反应物的选择规则,另一个是产物的生成规则。反应物选择规则是对反应系统中可能发生该步反应的各种反应物分子集合;产物生成规则是反应物分子集合发生该反应后的产物分子集合。
针对单环环烷烃,制定了包括β-裂化、异构化、开环、脱氢环化、氢转移、聚合等11大类共计37条反应规则。如,允许质子进攻分子的C—C、C—H键以及烷基取代苯侧链C—C键;禁止甲基碳正离子和质子的形成,没有甲基、乙基碳正离子生成,不会同时生成多个碳正离子,生成碳正离子顺序按叔、仲、伯进行;缩环反应需要分子结构中有六元环烷环;扩环反应需要分子结构中有五元环烷环;环烷烃与烯烃之间的氢转移反应都以生成具有芳烃结构结束;不考虑热裂化反应等。
3.4 反应网络生成
以催化裂化反应化学为指导,结合制定的37条反应规则,建立了基于反应路径层面兼顾机理层面的OCHA催化裂化反应网络。该反应网络包含216个化学反应,分子数为71。OCHA催化裂化各反应族的反应数目列于表3。由表3可知,裂化反应数量较多,达到107个,与实验中得到的取代环烷烃催化裂化反应规律相一致。
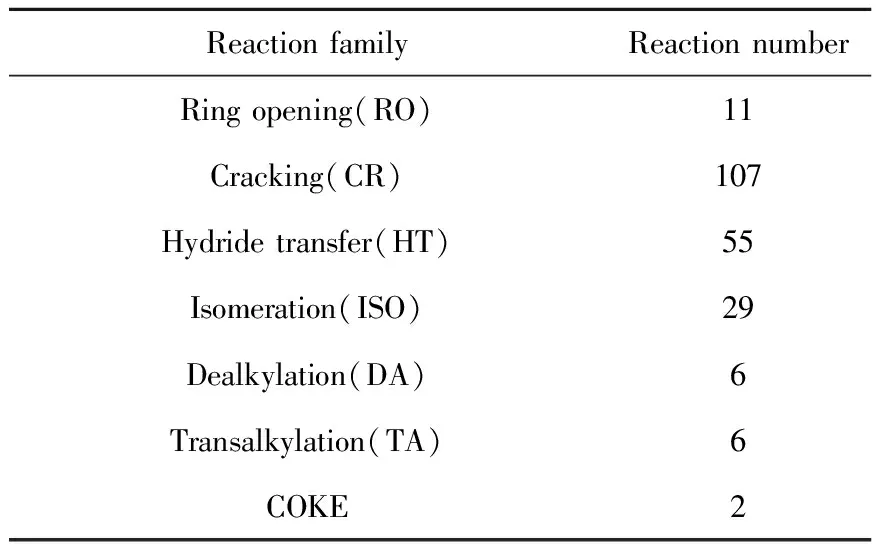
表3 OCHA催化裂化反应各反应族的反应数目
笔者在前期的研究工作中建立了求取分子水平动力学参数初值的方法,得到了甲基环己烷(MCHA)催化裂化反应中的指前因子数值[15-16],可以作为本模型计算的初值。
3.6 动力学模型求解
以A→B+C反应为例,建立动力学模型。式(1)~式(4)分别表示模型中的基元反应速率、反应速率常数、组分浓度及每一类反应的速率常数表达式。
A→B+C
(1)
k=[kcatalytic,x…kcatalytic,y…]T
(2)
C=[Cbulk,xiCbulk,xj…Ccations,xiCcations,xj…]
(3)
(4)
当搭建好反应网络、获得模型中所需的动力学参数初值后,运用Levenberg-Marquardt(LM)算法对建立的方程进行回归计算。首先,以一定时间步长Δt对产物分布进行模拟计算;随后,比较预测值和实验值,若方差达到设定的收敛精度,则停止计算,否则进一步调整动力学参数值,直到满足设定的收敛精度为止。
4 OCHA催化裂化反应动力学模型预测结果与实验结果的比较
4.1 模型预测结果与实验结果的比较
在6组实验数据中,选取转化率为9.9%、23.3%、32.8%、60.4%的4组数据回归动力学参数,剩余2组数据进行验证。图1为烷烃、烯烃、环烷烃、芳烃和焦炭5大类产物模型计算值与实验值对比图。由图1可知,对于丙烯、丁烯、戊烯、己烯、庚烯等烯烃类以及C7、C8环烷烃的产物预测较为准确。
由于实验分析的限制,部分产物根据结构相似性原则进行了拆分、合并处理,且反应网络中的分子数量多于实验分析数量,在一定程度上也给模型计算带来了额外的误差。另外,从实验分析的角度,应尽可能对每一产物分子进行鉴别。虽然与Watson等[14]建立的OCHA机理水平催化裂化反应动力学模型精度有一定的差距,他们建立的模型预测值与实验值误差在5%内,但是笔者建立的反应路径层面动力学模型不需要大量的理论计算,且模型建立过程较为简单、扩展性较好,动力学参数可用于其他催化裂化反应体系。
傅江峰死后,其子傅晓渊思念不已,向友人永嘉人士胡琴舟诉说在梅岭的读书生活。胡琴舟感念其父子情深,根据他的叙述,于光绪十六年(1890)精心绘制了一幅傅岱结庐教子图,再现了当年其父授课情景。傅晓渊见图,如获至宝,随即命名为《梅岭课子图》,图名请老师俞樾题写。
从模拟结果来看,各转化率下的模型预测值和实验值吻合良好,只有异构烷烃、正构烷烃以及多取代的烷烃、芳烃等几种产物偏差较大。OCHA催化裂化生成异构烷烃的主要途径是OCHA发生侧链断裂,生成的小分子烯烃和环烷烃再发生氢转移反应。而二取代的环烷烃、芳烃主要由烷基转移得到。由于在模型中有很多的中间体物种,在实际的产物中含量为零,故而很难准确预测,最终导致异构烃类、多取代的环烷烃和芳烃产物含量预测值与实验值的差别。这与笔者在建立MCHA分子水平动力学模型得到的结论相类似。
表4给出了OCHA催化裂化反应的16类反应速率常数(前4组优化得到的速率常数平均值)。将优化得到的动力学参数与先前工作中得到的初值[15-16]进行对比,发现两者之间数量级无大的差别,仅个别数据的差别较大(如脱烷基反应),说明其初值具有较好的准确性。活化能常数项E0除了反映催化剂的不同,还可以比较不同反应族之间发生反应的难易程度[18-19]。由Arrhenius公式可知,反应的速率常数k值越大,活化能越低,该反应越容易发生[17]。正构烯烃的异构化、β-裂化、氢转移反应的k值较其他反应类型数值大,说明了该类反应易于发生,与实验现象吻合。
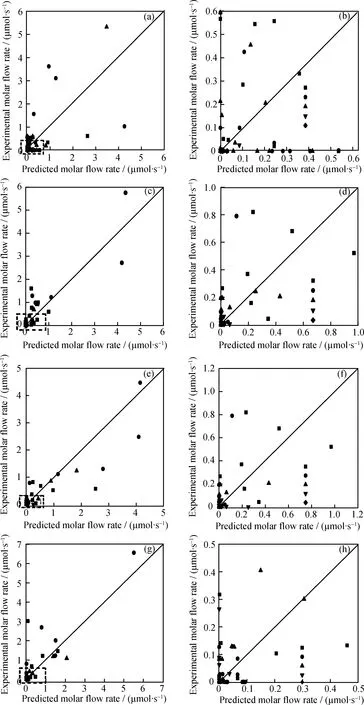
图1 OCHA催化裂化反应不同转化率下主要产物摩尔流率的模拟值与实验值比较
表4 回归得到的OCHA催化裂化动力学参数
Table 4 Optimized kinetic parameters for OCHA catalytic cracking
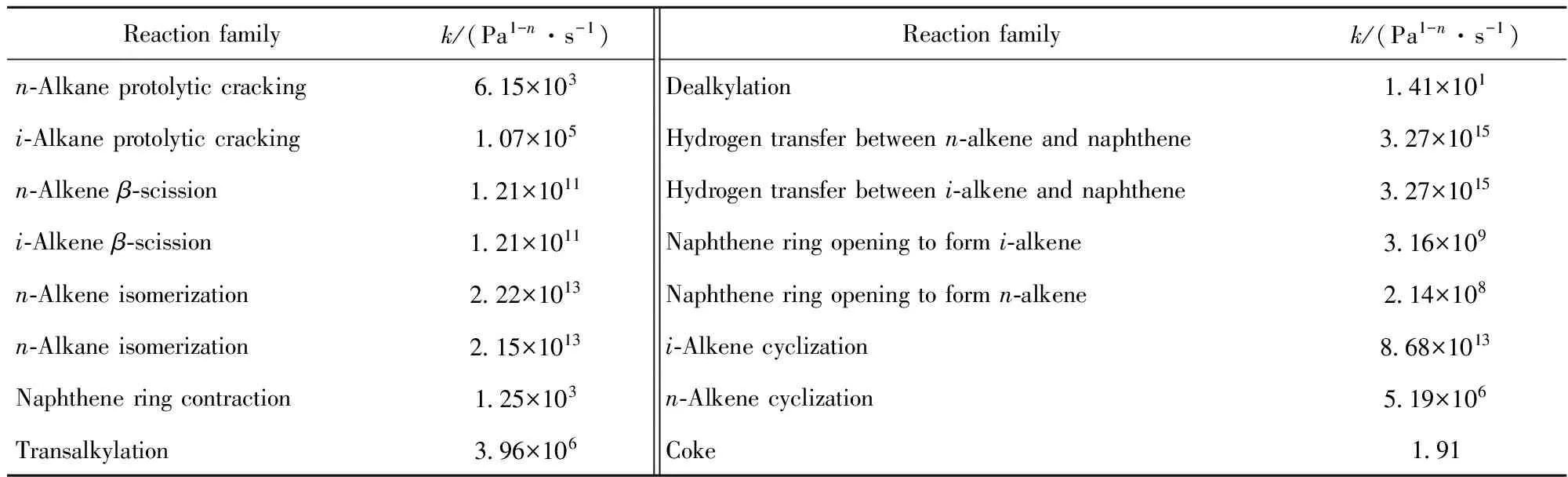
Reactionfamilyk/(Pa1-n·s-1)Reactionfamilyk/(Pa1-n·s-1)n⁃Alkaneprotolyticcracking6 15×103Dealkylation1 41×101i⁃Alkaneprotolyticcracking1 07×105Hydrogentransferbetweenn⁃alkeneandnaphthene3 27×1015n⁃Alkeneβ⁃scission1 21×1011Hydrogentransferbetweeni⁃alkeneandnaphthene3 27×1015i⁃Alkeneβ⁃scission1 21×1011Naphtheneringopeningtoformi⁃alkene3 16×109n⁃Alkeneisomerization2 22×1013Naphtheneringopeningtoformn⁃alkene2 14×108n⁃Alkaneisomerization2 15×1013i⁃Alkenecyclization8 68×1013Naphtheneringcontraction1 25×103n⁃Alkenecyclization5 19×106Transalkylation3 96×106Coke1 91
T=823 K
4.2 模型验证结果与实验结果的比较
为了进一步验证模型的适应性与准确性,选取了OCHA转化率为41.0%、65.0% 2组实验数据进行模型验证,结果示于图2,其相关系数达到了0.96、0.92,预测效果较好,说明模拟计算得到的动力学参数可以预测REY分子筛催化下的催化裂化反应。另外,经过MCHA、乙基环己烷(ECHA)等单环环烷烃的催化裂化实验数据验证,回归得到的16类动力学参数值也可以用于其动力学模型的建立,这也为进一步建立环烷烃催化裂化反应分子水平动力学模型提供数据支持。
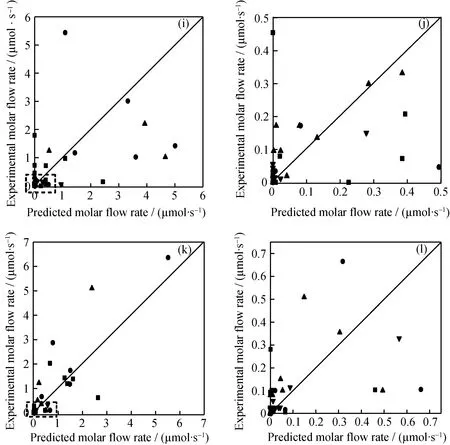
图2 OCHA催化裂化反应转化率为41.0%、65.0% 时主要产物摩尔流率实验数据的模型验证
随着反应物空速的降低,OCHA转化率由3751 h-1时的9.9%上升到120 h-1时的65.0%。图3是4种反应类型对OCHA转化率贡献大小的示意图。4种反应类型分别是,(1)质子进攻OCHA分子叔碳原子的C—H键;(2)质子进攻OCHA分子六元环上的C—C键;(3)质子进攻侧链C—C键;(4)氢转移反应。从图3可以看出,反应类型(1)在不同转化率下所占比例较小,基本可以忽略。在较低转化率情况下,反应类型(2)引发的OCHA开环占到OCHA总转化率的近90%,当OCHA转化率达到65.0%时,其所占比例下降到30%左右。反应类型(3)随转化率的变化幅度不大,其中质子进攻环上C—C键的几率大约在70%~85%之间,因为进攻环上C—C键时容易形成更加稳定的C14正碳离子,而进攻侧链上C—C键将会形成不稳定的低碳数正碳离子[14]。随着转化率的上升,反应类型(4)所占的比例由10%增加到60%左右,产物中烷烃与烯烃比以及芳烃的质量分数也随之增大。
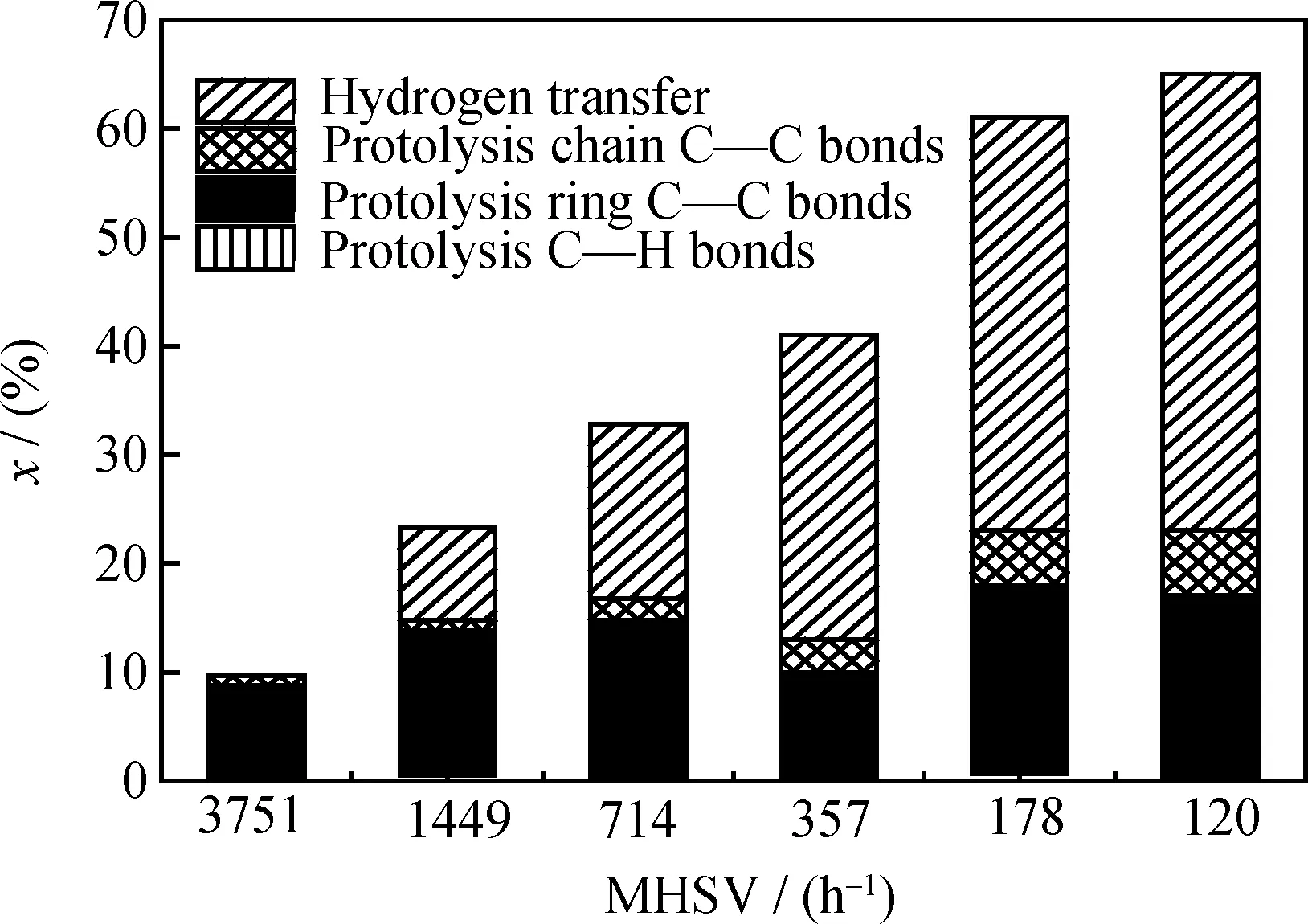
图3 各反应类型对OCHA催化裂化反应转化率贡献大小示意图
4.3 单环环烷烃转化规律
单环环烷烃催化裂化主反应包括环烷环的开环与随后的二次裂化反应。OCHA由于有长侧链,在裂化反应中侧链会发生异构化和氢转移反应。相对于开环、裂化和异构化反应,短取代基的MCHA比OCHA更容易发生氢转移反应。从产物分析也得到印证,OCHA的反应产物中有焦炭生成,而MCHA没有焦炭生成[15]。随着单环环烷烃取代基链长的增加,其转化率也会随之增大。OCHA结构比较稳定,不容易发生开环或芳构化反应,即使采用氢转移活性较高的Y型分子筛催化剂,其芳构化选择性也低于10%,因此单环环烷环结构的供氢性能不高,对生成低碳烯烃的影响不大。
5 结 论
通过反应原料和产物的分子管理、反应规则、动力学参数获取以及模型建立与求解,建立了基于OCHA模型化合物的单环环烷烃分子水平催化裂化动力学模型。制定了11大类共37条反应规则。随机选取了4组OCHA催化裂化实验数据进行了模拟计算,主要反应产物预测值与实验值吻合良好。回归得到了16类动力学参数值,将得到的动力学参数用于另外2组实验数据的验证,结果显示其预测能力良好,模型预测值与实验值的相关系数分别达到了0.96、0.92,说明模型具有较高的准确性。回归得到的16类动力学参数值可以用于建立其他单环环烷烃模型化合物分子水平动力学模型。另外,模型还可以用来预测催化裂化反应中氢转移反应的贡献大小。在低转化率条件下,质子氢进攻C—C键作为主要的反应途径;随着转化率的升高,氢转移反应水平不断增加,链烷烃/烯烃比与芳烃产物收率也随之增大。
符号说明:
C——组分浓度,mol/g;
Cbulk,xi——i物质在x类反应中的浓度,mol/g;
Cbulk,xj——j物质在x类反应中的浓度,mol/g;
Ccations,xi——i物质在x类反应中的离子浓度,mol/g;
Ccations,xj——j物质在x类反应中的离子浓度,mol/g;
Ea,x——x类反应活化能,J/mol;
h——Planck常数,6.62×10-34(J·s);
k——反应速率常数,速率系数的单位取决于反应的总级数;
kB——Boltzmann常数,1.38×10-23(J/K);
kcatalytic,x——x类反应的速率常数;
kcatalytic,y——y类反应的速率常数;
R——摩尔气体常数,8.314 J/(K·mol);
t——反应时间,s;
T——反应温度,K;
Δn——过渡态与反应物之间的分子数变化,单分子反应其值为0,双分子反应为-1;
ΔS0*——气相活化熵差,J/(mol·K);
[1] STANISLSUS A, MARAFI A, RAN M S. Recent advances in the science and technology of ultra low sulfur diesel (ULSD) production[J]. Catal Today, 2010, 153(1-2): 1-68.
[2] 张旭, 郭锦标, 周祥, 等. 分子水平动力学模型在催化裂化反应中的应用[J]. 化工进展, 2012, 31(12): 2678-2685. (ZHANG Xu, GUO Jinbiao, ZHOU Xiang, et al. Application of molecular level kinetic modeling on catalytic cracking reaction[J]. Chem Ind Eng Prog, 2012, 31(12): 2678-2685.)
[3] 张结喜, 齐艳华, 邱建章. 催化裂化关联模型的研究[J]. 计算机与应用化学, 2007, 24(11): 1519-1522. (ZHANG Jiexi, QI Yanhua, QIU Jianzhang. Study on FCC correlation models[J]. Comput Appl Chem, 2007, 24(11): 1519-1522.)
[4] 祝然, 沈本贤, 刘纪昌. 基于结构导向集总方法考察减压蜡油掺炼地沟油催化裂化效果[J]. 石油学报(石油加工), 2014, 30(3): 484-492. (ZHU Ran, SHEN Benxian, LIU Jichang. Effect of catalytic cracking of vacuum gas oil blended with waste oil based on structure oriented lumping method[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2014, 30(3): 484-492.)
[5] 江洪波, 钟贵江, 宁汇, 等. 重油催化裂化MIP工艺集总动力学模型[J]. 石油学报(石油加工), 2010, 26(6): 901-909. (JIANG Hongbo, ZHONG Guijiang, Ning Hui, et al. Lumping kinetic model of heavy oil catalytic cracking for MIP technology[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2010, 26(6): 901-909.)
[6] 许友好, 龚剑波, 张久顺, 等. 多产异构烷烃的催化裂化工艺两个反应区概念实验研究[J]. 石油学报(石油加工), 2004, 20(4): 1-5. (XU Youhao, GONG Jianhong, ZHANG Jiushun, et al. Experimental study on “two reaction zone” conception connected with MIP process[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2004, 20(4): 1-5.)
[7] 王建平, 许先焜, 翁惠新, 等. 加氢渣油催化裂化14集总动力学模型的建立[J]. 化工学报, 2007, 58(1): 86-94. (WANG Jianping, XU Xiankun, WENG Huixin, et al. Establishment of 14 lumps model for fluid catalytic cracking of hydrotreated residuum[J]. J Chem Ind Eng, 2007, 58(1): 86-94.)
[9] WEI W, BENNETT C A, TANAKA R, et al. Computer aided kinetic modeling with KMT and KME[J]. Fuel Process Technol, 2008, 89(4): 350-363.
[10] JAFFE S B, FREUND H, OLMSTEAD W N. Extension of structure-oriented lumping to vacuum residua[J]. Ind Eng Chem Res, 2005, 44(26): 9840-9852.
[11] WATSON B A, KLEIN M T, HARDING R H. Mechanistic modeling ofn-heptane cracking on HZSM-5[J]. Ind Eng Chem Res, 1996, 35(5): 1506-1516.
[12] WATSON B A, KLEIN M T, HARDING R H. Mechanistic modeling of n-hexadecane cracking on rare earth Y[J]. Energ Fuels, 1997, 11(2): 354-363.
[13] WATSON B A, KLEIN M T, HARDING R H. Mechanistic modeling of a 1-phenyloctane/n-hexadecane mixture on rare earth Y zeolite[J]. Ind Eng Chem Res, 1997, 36(8): 2954-2963.
[14] WATSON B A, KLEIN M T, HARDING R H. Catalytic cracking of alkylcyclohexanes: Modeling the reaction pathways and mechanisms[J]. Int J Chem Kinet, 1997, 29(7): 545-560.
[15] 张旭, 郭锦标, 周祥, 等. 甲基环己烷催化裂化分子水平反应动力学模型的建立[J]. 石油学报(石油加工), 2014, 30(5): 829-836. (ZHANG Xu, GUO Jinbiao, ZHOU Xiang, et al. Establishment of molecular level kinetic model for methylcyclohexane catalytic cracking[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2014, 30(5): 829-836.)
[16] 张旭, 郭锦标, 周祥, 等. 单环环烷烃催化裂化动力学模型的建立-指前因子的计算[J]. 石油学报(石油加工), 2013, 29(2): 283-288. (ZHANG Xu, GUO Jinbiao, ZHOU Xiang, et al. Kinetic modeling of catalytic cracking of monocyclic cycloparaffins—Calculation of pre-exponential factors[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2013, 29(2): 283-288.)
[17] 杜梅西克, 拉德, 阿帕里西奥, 等. 多相催化微观动力学(沈俭一译)[M]. 北京: 国防工业出版社, 1998: 25-40.
[18] AL-SABAWI M N, DE LASA H. Kinetic modeling of catalytic conversion of methylcyclohexane over USY zeolites: Adsorption and reaction phenomena[J]. AIChE J, 2009, 55(6): 1538-1558.
[19] AL-SABAWI M N. Heterogeneous kinetic modeling of the catalytic conversion of cycloparaffins[D]. Ontario: The University of Western Ontario, 2009.
Molecular-Based Construction of Kinetic Models for Monocycloparafffin Catalytic Cracking
ZHANG Xu, ZHOU Xiang, WANG Xinlei, GUO Jinbiao
(ResearchInstituteofPetroleumProcessing,SINOPEC,Beijing100083,China)
Based on the catalytic cracking mechanism of cycloparaffins, 37 reaction rules for monocyclic cycloparaffins catalytic cracking were formulated according to reaction family conception. A molecular level kinetic model was established for monocycloparafffins catalytic cracking by means of quantum chemical calculations combined with reaction family method. The kinetic model was verified by a model compound of octylcyclohexane, and kinetic parameter values for sixteen types of octylcyclohexane catalytic cracking were obtained. The simulation results showed that the predicted molar flow rate values of main reaction products were in good agreement with the experimental values, and the contribution values of four main types of reactions to the conversion of octylcyclohexane catalytic cracking were obtained. In addition, the kinetic parameters could be used for the catalytic cracking reaction of other monocyclic cycloparaffins, and further for establishment of cracking reaction kinetic model of other cycloparaffins.
molecular level; kinetic model; kinetic parameters; monocycloparafffin; catalytic cracking
2014-11-25
张旭,男,博士,从事催化裂化反应动力学方面的研究
郭锦标,男,教授级高级工程师,博士,从事计算机在石油化工生产、经营和管理中的应用研究工作;Tel: 010-82368632;E-mail:guojinbiao.ripp@sinopec.com
1001-8719(2015)06-1345-09
TE624.4
A
10.3969/j.issn.1001-8719.2015.06.014
猜你喜欢
石油炼制与化工(2022年6期)2022-06-21
食品研究与开发(2022年1期)2022-01-24
水产学杂志(2021年4期)2021-10-18
煤炭转化(2020年2期)2020-04-24
石油石化绿色低碳(2019年6期)2019-01-14
石油石化绿色低碳(2019年6期)2019-01-14
润滑油(2016年4期)2016-11-04
公民与法治(2016年24期)2016-05-17
当代化工研究(2016年6期)2016-03-20
化工进展(2015年6期)2015-11-13
