
Cr/TiO2 催化剂汞氧化的实验
2015-07-11 10:09俞晋频邱坤赞周劲松
浙江大学学报(工学版) 2015年11期
俞晋频,邱坤赞,宋 浩,高 翔,周劲松
(浙江大学 能源清洁利用国家重点实验室,浙江 杭州310027)
汞作为大气污染物之一,由于具有极强的神经毒性、生物累积性和可迁移性,已经受到越来越大的重视[1].燃煤电厂是人为汞排放的主要来源之一.根据联合国环境规划署(UNEP)2013 年的报告[2],2010年因为燃煤向大气中排放大约474t汞,约占人为汞排放总量的24%.2011年12月,美国环境保护署(EPA)颁布了首个全国性的汞和有毒空气污染物排放标准(mercury and air toxics standards,MATS)限制燃煤电厂汞排放[3].全球范围内汞排放标准越来越严格是必然趋势,因此亟需高效而且经济的脱汞技术.
在电厂燃煤过程中,煤中的汞释放到烟气中,经过一系列物理化学转化过程,最终以3种形式存在:零价汞(Hg0)、二价汞(Hg2+)和颗粒汞(Hgp)[4].其中颗粒汞可以在除尘装置(ESP 或FF)中脱除,二价汞易于在湿法脱硫装置(WFGD)中脱除,只有零价汞难以在现有的污染物控制装备中直接脱除.现在燃煤电厂还没有一套成熟高效可供推广的脱汞技术.活性炭喷射是目前最接近实际应用的脱汞技术[5].但是活性炭喷射成本较高,需要增加额外设备,并且吸附的活性炭需要处理.考虑到现在多数电厂配备了脱硝、除尘和脱硫等污染物控制设备,催化氧化是一种具有较大前景的脱汞技术.通过催化剂将零价汞氧化成二价汞,然后在后继的污染物控制设备中加以脱除,可以极大地降低污染物控制成本.
国内外学者研究了过渡金属的氧化物或化合物单独或负载在不同载体上用于汞氧化的可能性,包括 Fe2O3/TiO[6]2、 MnOx/TiO[7,28]、 Co/TiO[9]2、CuCl2/TiO[10]2等.考虑到Cr氧化物良好的氧化能力,Cr/TiO2催化剂也被尝试用于汞氧化.此外Cr氧化物作为活性组分可以催化Deacon 制氯反应[11-12],而氯气的产生在汞氧化过程中至关重要.因此本文以TiO2为载体,负载Cr的氧化物,考察其汞氧化能力.
本文用等体积浸渍法制备了不同质量分数的Cr/TiO2催化剂,通过X 射线衍射和N2物理吸附研究了催化剂的表面结构特性,重点研究了Cr负载量、反应温度、不同烟气组分对Cr/TiO2催化剂上汞氧化的影响.
1 实验部分
1.1 实验装置
汞氧化活性实验台如图1所示.该实验台主要由配气系统、固定床反应器和汞浓度监测仪3部分组成.模拟烟气由N2、O2、HCl、NO、SO2、H2O 和Hg0组成.N2、O2、HCl、NO 和SO2由钢瓶气提供,并由质量流量计精确控制流量.液态H2O 通过蠕动泵精确控制流量,汽化成水蒸气后与其他气体混合.在汞发生器中,渗透管在恒定温度和恒定载气流量下产生特定浓度的汞蒸气.固定床反应器由程序温度控制仪精确控制温度.反应器出口气体通过汞浓度监测仪即时检测浓度,尾气经处理后排空.实验台有Hg0通过的管路均有伴热带加热,防止Hg0沉积在管路上影响测量.

图1 汞氧化活性实验台示意图Fig.1 Schematic diagram of mercury oxidation activity test reactor system
1.2 Cr/TiO2 催化剂的制备和表征
本文研究的Cr/TiO2催化剂用等体积浸渍法制备得到.载体采用锐钛矿TiO2,前驱体为不同浓度的硝酸铬溶液.TiO2载体使用前在120°C 空气气氛下干燥脱水4h.称取一定量的TiO2,按照TiO2的吸水率加入特定体积的硝酸铬溶液.用玻璃棒充分搅拌均匀后,室温静置过夜.在120°C 空气气氛下干燥12h,然后在500°C 空气气氛下煅烧5 h.制备得到质量分数分别为0.1%、0.5%、1%、2%和5%的Cr/TiO2催化剂,研磨成颗粒用作表征和活性测试.
催化剂结晶形态分析采用荷兰帕纳科X’Pert PRO X 射线衍射仪,管电压40KV,管电流40mA,Cu靶Kα 射线(λ=0.154nm),扫描角度为5°~80°,扫描步长为0.0167°,扫描速率为1°/min.
催化剂比表面积和孔分布由TRISTAR II3020全自动比表面和孔隙分析仪(美国Micromeritics Instrument Corporation制造)通过静态N2物理吸附法测定,吸附温度为77K.样品测试前,在300°C条件下真空脱气4h.比表面积通过BET 方程计算,孔径和孔容采用BJH 方程通过N2吸附等温线的脱附曲线计算.
1.3 汞氧化性能测试方法
在石英管中装填一定量的催化剂,用石英棉固定位置.程序控制预热单元和反应器达到特定温度.携带Hg0的模拟烟气首先经过旁路,浓度稳定后记录下初始值.然后切换模拟烟气经过催化剂,改变工况进行实验.催化剂出口汞浓度稳定后记录下数值.实验中Hg0氧化率定义为

2 结果与讨论
2.1 Cr/TiO2催化剂表征
不同含量Cr/TiO2催化剂的XRD 结果如图2所示.从图中可以看出,在TiO2载体和Cr/TiO2催化剂中只检测到锐钛矿型TiO2.0.1%~5%的Cr/TiO2催化剂中均没有检测到铬氧化物的晶型,说明在这些催化剂上铬氧化物呈无定型状态.
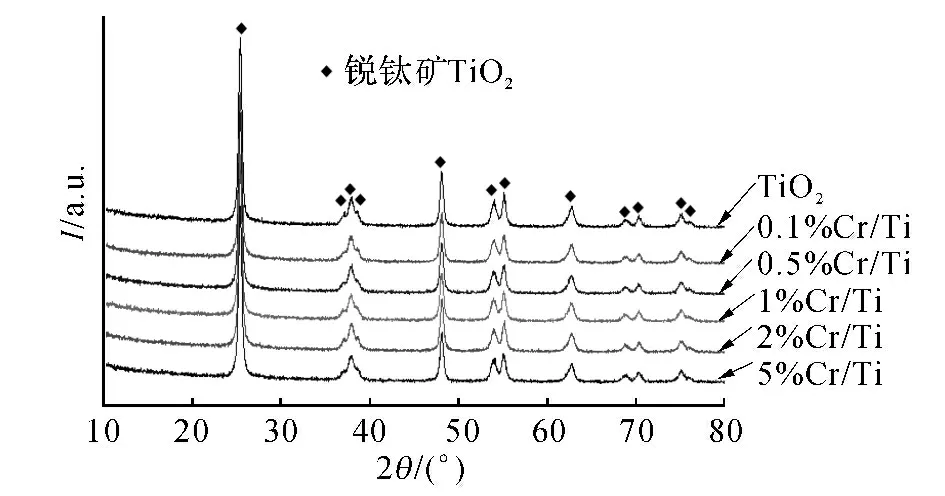
图2 不同含量Cr/TiO2 催化剂X射线衍射图谱Fig.2 X-ray diffraction patterns for Cr/TiO2catalysts containing different amounts of chromium
如表1所示显示了不同Cr含量催化剂的表面结构性质.表中S 为比表面积,V 为孔容,d 为孔径.从表中可以看出,Cr的负载对催化剂比表面积影响不大,该系列催化剂比表面积均在80m2·g-1以上.催化剂孔径随负载量提高呈降低趋势,这应该是由于负载的Cr氧化物堵塞了部分小孔.催化剂孔容没有随负载量变化呈现出特定的规律.

表1 不同含量Cr/TiO2 催化剂物理特性Tab.1 Physical characteristics of Cr/TiO2catalysts containing different amounts of chromium
2.2 Cr/Ti催化剂汞氧化性能
不同质量分数的Cr/TiO2催化剂在200~400°C范围内的汞氧化率如图3所示.实验条件为催化剂0.05g,总流量2L/min,Hg0初始浓度ρ(Hg0)约为85μg/m3,加入5%O2、5×10-6HCl、300×10-6NO 和500×10-6SO2.从图中可以看出,未负载Cr的TiO2对零价汞的氧化作用较小,氧化率在10%以下.随着Cr含量的提高,Cr/TiO2催化剂汞氧化率明显提高.0.5%的Cr/TiO2催化剂已经具有较好的汞氧化效果,350°C 时汞氧化率达到50.6%.当Cr含量提高到1%时,整个温度范围内汞氧化率在50%以上,350°C 时汞氧化率达到85.1%.5%的Cr/TiO2催化剂在整个温度范围内汞氧化率均超过95%.对每个催化剂而言,随着反应温度的升高,活化分子数目增多,汞氧化率随之提高.一些文献中报道随着反应温度的提高,零价汞在催化剂表面的吸附减少,汞氧化率有一定程度的降低[13-15].对Cr/TiO2催化剂而言,实验温度范围内没有观察到这种现象,反应温度对分子活化能的影响超过了对Hg0吸附的影响.

图3 不同含量Cr/TiO2 催化剂Hg0 氧化率Fig.3 Mercury oxidation of Cr/TiO2 catalysts containing different amounts of chromium
2.3 烟气成分对汞氧化的影响
为了研究不同烟气组分对Cr/TiO2催化剂上汞氧化的影响,选取了质量分数为1%的Cr/TiO2催化剂,在不同烟气条件下进行了汞氧化实验,结果如图4~11所示.实验条件为催化剂0.05g,总流量2L/min,温度350 ℃,Hg0初始浓度约为85μg/m3.从图4可以看出,在N2气氛下,Cr/TiO2催化剂对零价汞基本没有氧化作用.添加1%的O2时,Hg0氧化率提高到11.1%.O2浓度提高到2%和5%时,Hg0氧化率达到21.1%和43.3%.为了探究只有O2存在时催化剂上汞氧化机理,在较长时间段内同时检测了催化剂出口Hg0和Hg2+的浓度,实验结果如图5所示.从图中可以看出,当Hg0从旁路切换到催化剂管路时,Hg0浓度迅速降低,但是随着时间t推移,Hg0浓度缓慢上升,最终恢复到接近初始浓度,这个过程中没有检测到Hg2+.说明反应刚开始时Hg0吸附到催化剂表面,与晶格氧反应生成HgO,HgO 吸附在催化剂表面,随着晶格氧逐渐消耗,Hg0氧化逐渐停止.加入O2后,Hg0浓度先降低,然后升高并逐渐稳定下来.并且随着反应的进行,出口Hg2+逐渐上升并达到稳定.说明有O2存在的情况下,消耗的晶格氧得到补充,生成的HgO在催化剂表面吸附饱和以后挥发到模拟烟气中.根据现阶段已经提出的反应机理,在只有O2的条件下,汞氧化可能按照Mars-Maessen机理[9,16]进行:Hg0吸附到催化剂表面,与晶格氧反应生成弱吸附的HgO,催化剂消耗掉的晶格氧由气态O2补充,保证反应进行下去,如反应式(2)、(3)和(4)所示.

图4 O2 对Cr/TiO2 催化剂汞氧化的影响Fig.4 Influence of oxygen on mercury oxidation over Cr/TiO2catalyst

图5 O2 存在条件下Hg0 和Hg2+浓度Fig.5 Concentration of Hg0and Hg2+when O2exists
烟气中氧化态汞主要以HgCl2形式存在,所以HCl是影响汞氧化的最重要因素[17].如图6所示为HCl对Cr/TiO2催化剂汞氧化的影响,从图6中可以看出,添加1×10-6HCl时,汞氧化率就达到30.6%.HCl浓度为10×10-6时,汞氧化率可以达到60.0%.只有HCl而没有O2存在时,HCl可能吸附到催化剂表面,与晶格氧反应生成活性氯物种,活性氯物种与Hg0反应完成汞氧化.有O2存在时,HCl对汞氧化的促进作用更为明显.在5%O2条件下,添加1×10-6、5×10-6和10×10-6HCl,汞氧化均在90%以上,实现了低氯条件下Hg0的高效氧化.1×10-6HCl+5%O2条件下Hg0氧化率大于单独添加1×10-6HCl和5%O2时的氧化率之和,这说明HCl和O2同时存在时汞氧化机理发生了改变.一方面,HCl和O2同时存在时,由于Cr氧化物对Deacon反应的催化作用,可能生成Cl2或Cl的中间物种,如反应式(5)所示,而这些物种对汞氧化有极强的促进作用;另一方面,HCl存在时弱吸附态的HgO 与活性氯进一步反应生成HgCl2并从催化剂表面脱附,如反应式(6)所示,这就促进了O2和Hg0之间的反应.另外,活性氯可能直接与气态Hg0反应,生成HgCl2,如反应式(7)所示.因此,Cr/TiO2催化剂在低氯条件下表现出很高的汞氧化活性.在HCl和O2同时存在的条件下,检测了催化剂出口Hg0和Hg2+的浓度,结果如图7所示.ρ(Hg)为质量浓度,从图中可以看出,当加入HCl和O2后,初始阶段Hg0有一个小的脱附,这可能是HCl与Hg0竞争吸附造成的.在这之后,Hg0浓度逐渐降低,Hg2+浓度逐渐升高,最终Hg0和Hg2+浓度都趋于稳定.说明Cr/TiO2催化剂上的汞氧化达到了平衡状态,Hg0浓度和Hg2+浓度之和略小于初始Hg0浓度,可能是有少量的氧化态汞残留在了催化剂表面.

如图8 所示为NO 对Cr/TiO2催化剂汞氧化的影响,从图8中可以看出,单独添加NO 时,对汞氧化基本没有影响.但是在O2存在的条件下,NO的添加可以促进汞氧化.相比5%O2条件下43.4%的氧化率,添加100×10-6或300×10-6的NO,汞氧化率分别提高到52.9%和58.8%.O2和HCl同时存在时,NO 的添加也可以促进汞氧化.其他一些研究者的工作中也观察到了NO 对汞氧化的促进作用[7,18-21].NO 促进汞氧化可能是由于NO 吸附到催化剂表面生成了NO+、NO2、NO-3等中间物种,这些中间物种起到了汞氧化的作用.

图6 HCl对Cr/TiO2 催化剂汞氧化的影响Fig.6 Influence of hydrogen chloride on mercury oxidation over Cr/TiO2catalyst

图7 O2 和HCl同时存在时Hg0 和Hg2+浓度Fig.7 Concentration of Hg0 and Hg2+when O2 and HCl exist

图8 NO 对Cr/TiO2 催化剂汞氧化的影响Fig.8 Influence of nitric oxide on mercury oxidation over Cr/TiO2catalyst
SO2对汞氧化的影响作用较为复杂,有些研究工作中发现SO2有促进作用[22-23],另有一些研究发现SO2起抑制作用[17,24-25]或基本没有影响[20].如图9所示为SO2对Cr/TiO2催化剂汞氧化的影响,从图中可以看出,单独添加SO2时,对汞氧化基本没有影响.有O2存在条件下,SO2对汞氧化有很强的抑制作用.添加100×10-6和500×10-6的SO2时,汞氧化率分别降低到9.4%和3.5%.在O2和HCl同时存在时,SO2也对汞氧化起抑制作用,添加100×10-6和500×10-6的SO2时,汞氧化率分别降低到65.9%和57.6%.SO2抑制Cr/TiO2催化剂上的汞氧化,其原因很可能是SO2与Hg0竞争吸附到催化剂表面[25].只有O2存在时,吸附态Hg0与晶格氧反应,SO2和Hg0竞争吸附到催化剂表面,因此对汞氧化抑制作用很强;O2和HCl同时存在时,部分气态Hg0直接与活性氯物种反应,因此受SO2竞争吸附的影响较小.

图9 SO2 对Cr/TiO2 催化剂汞氧化的影响Fig.9 Influence of sulfur dioxide on mercury oxidation over Cr/TiO2catalyst
为探究SO2对Cr/TiO2催化剂上汞氧化的抑制机理,在1%Cr/TiO2催化剂上进行了Hg0吸附和脱附的实验,如图10所示.催化剂用量为0.2g,总流量2L/min,Hg0初始浓度约为85μg/m3.催化剂首先在200°C、Hg0/N2气氛下吸附饱和,然后停止通Hg0,分别用N2和SO2/N2吹扫.从图中可以看出,用N2吹扫时,Hg0浓度很快降低到零.用SO2/N2吹扫时,观察到Hg0浓度先上升,然后缓慢降低到零.在这种情况下,SO2使Hg0从催化剂表面脱附.说明SO2和Hg0在催化剂表面有相同的吸附位点,并且在此吸附位点上SO2的吸附能力强于Hg0.
如图11所示显示了H2O 对Cr/TiO2催化剂上汞氧化的影响,从图中可以看出H2O 对汞氧化有较大的抑制作用.模拟烟气(SFG:5%O2,10×10-6HCl,300×10-6NO,500×10-6SO2)条件下1%Cr/TiO2催化剂有89.8%的汞氧化率,添加1%H2O 时汞氧化率降到72.7%,添加5%H2O 时汞氧化率降到40.9%.其他种类的催化剂上也观察到了H2O 的 抑 制 作 用[26,27],H2O 对 汞 氧 化 的 抑 制 可 能是由于H2O 抑制了Hg0在催化剂表面的吸附.需要指出的是,为了体现不同烟气成分的影响,实验中空速设置较高,为960 000h-1.实际应用中空速较小,因此会有更高的汞氧化率.

图10 SO2 促进1%Cr/TiO2 催化剂上汞的脱附Fig.10 Mercury desorption over 1%Cr/TiO2promoted by SO2

图11 H2O 对Cr/TiO2 催化剂汞氧化的影响Fig.11 Influence of water vapor on mercury oxidation over Cr/TiO2catalyst
3 结 论
用等体积浸渍法制备了不同质量分数(0.1%~5%)的Cr/TiO2催化剂,X 射线衍射和N2吸附结果显示铬氧化物在催化剂表面呈无定型状态,并且催化剂有较高的比表面积,活性测试表明Cr/TiO2催化剂在高空速、低氯条件下有很高的汞氧化活性.
随着Cr含量的提高,Cr/TiO2催化剂汞氧化活性明显提高,并且在实验温度范围内(200~400°C),汞氧化率随温度升高而提高.
O2和HCl对汞氧化有促进作用.HCl存在条件下,Cr/TiO2催化剂表面生成了较多活性氯,因此有很高的汞氧化率.
NO 也可以促进汞氧化,其原因可能是在催化剂表面生成了NO2等中间物种,与Hg0反应生成HgO.
SO2对汞氧化有抑制作用.SO2与Hg0在催化剂表面有竞争吸附的关系,可以导致Hg0从催化剂表面脱附.H2O 也会抑制汞氧化.
(Reference):
[1]REDDY B,DURGASRI N,KUMAR T,et al.Abatement of gas-phase mercury—recent developments[J].Catalysis Reviews,2012,54:344-398.
[2]United Nations Environment Programme.Global mercury assessment 2013:sources,emissions,releases and environmental transport[R].Geneva:Switzerland UNEP Chemicals Branch,2013.
[3]United States Environmental Protection Agency.Mercury and air toxics standards(MATS)[EB/OL].(2015-9-25)[2015-10-7].http://www.epa.gov/airquality/powerplanttoxics/actions.html.
[4]GALBREATH K,ZYGARLICKE C.Mercury transformations in coal combustion flue gas[J].Fuel Processing Technology,2000,65:289-310.
[5]PAVLISH J,SONDREAL E,MANN M,et al.Status review of mercury control options for coal-fired power plants[J].Fuel Processing Technology,2003,82:89-165.
[6]WU Sheng-ji,OZAKI M,UDDIN M,et al.Development of iron-based sorbents for Hg0removal from coal derived fuel gas:Effect of hydrogen chloride[J].Fuel,2008,87:467-474.
[7]JI Lei,SREEKANTH P,SMIRNIOTIS P,et al.Manganese oxide/titania materials for removal of NOxand elemental mercury from flue gas[J].Energy & Fuels,2008,22:2299-2306.
[8]XIE Jiang-kun,YAN Nai-qiang,YANG Shi-jian,et al.Synthesis and characterization of nano-sized Mn–TiO2catalysts and their application to removal of gaseous elemental mercury[J].Research on Chemical Intermediates,2012,38:2511-2522.
[9]LIU Yue,WANG Yue-jun,WANG Hai-qiang,et al.Catalytic oxidation of gas-phase mercury over Co/TiO2catalysts prepared by sol–gel method[J].Catalysis Communications,2011,12:1291-1294.
[10]KIM M,HAM S,LEE J.Oxidation of gaseous elemental mercury by hydrochloric acid over CuCl2/TiO2-based catalysts in SCR process[J].Applied Catalysis B:Environmental,2010,99:272-278.
[11]AGLULIN A.Kinetics and possible mechanism of HCl oxidation on chromium-containing catalysts[J].Kinetics and Catalysis,1998,39:521-529.
[12]AMRUTE A,MONDELLI C,PEREZ-RAMIREZ J.Kinetic aspects and deactivation behaviour of chromiabased catalysts in hydrogen chloride oxidation[J].Ca-talysis Science &Technology,2012,2:2057-2065.
[13]STRAUBE S,HAHN T,KOESER H.Adsorption and oxidation of mercury in tail-end SCR-DeNOxplants—Bench scale investigations and speciation experiments[J].Applied Catalysis B:Environmental,2008,79:286-295.
[14]LEE C,SERRE S,ZHAO Y,et al.Mercury oxidation promoted by a selective catalytic reduction catalyst under simulated Powder River Basin coal combustion conditions[J].Journal of the Air & Waste Management Association,2008,58:484-493.
[15]RALLO M,HEIDEL B,BRECHTEL K,et al.Effect of SCR operation variables on mercury speciation[J].Chemical Engineering Journal,2012,198-199:87-94.
[16]GRANITE E,PENNLINE H,HARGIS R.Novel sorbents for mercury removal from flue gas[J].Industrial &Engineering Chemistry Research,2000,39:1020-1029.
[17]CAO Yan,CHEN Bobby,WU Jiang,et al.Study of mercury oxidation by a selective catalytic reduction catalyst in a pilot-scale slipstream reactor at a utility boiler burning bituminous coal[J].Energy &Fuels,2007,21:145-156.
[18]FAN Xiao-peng,LI Cai-ting,ZENG Guang-ming,et al.Hg0Removal from simulated flue gas over CeO2/HZSM-5[J].Energy &Fuels,2012,26:2082-2089.
[19]LI Hai-long,WU Chang-Yu,LI Ying,et al.CeO2-TiO2Catalysts for catalytic oxidation of elemental mercury in low-rank coal combustion flue gas[J].Environmental Science &Technology,2011,45:7394-7400.
[20]LI Ying,MURPHY P,WU Chang-Yu,et al.Development of silica/vanadia/titania catalysts for removal of elemental mercury from coal-combustion flue gas[J].Environmental Science & Technology,2008,42:5304-5309.
[21]LI Ying,MURPHY P,WU Chang-Yu.Removal of elemental mercury from simulated coal-combustion flue gas using a SiO2–TiO2nanocomposite[J].Fuel Processing Technology,2008,89:567-573.
[22]ESWARAN S,STENGER H.Understanding mercury conversion in selective catalytic reduction(SCR)catalysts[J].Energy &Fuels,2005,19:2328-2334.
[23]FAN Xiao-peng,LI Cai-ting,ZENG Guang-ming,et al.Removal of gas-phase element mercury by activated carbon fiber impregnated with CeO2[J].Energy &Fuels,2010,24:4250-4254.
[24]WEN Xiao-yu,LI Cai-ting,FAN Xiao-peng,et al.Experimental study of gaseous elemental mercury removal with CeO2/gamma-Al2O3[J].Energy & Fuels,2011,25:2939-2944.
[25]ZHUANG Ye,LAUMB J,LIGGETT R,et al.Impacts of acid gases on mercury oxidation across SCR catalyst[J].Fuel Processing Technology,2007,88:929-934.
[26]LI Hai-long,WU Chang-Yu,LI Ying,et al.Role of flue gas components in mercury oxidation over TiO2supported MnOx-CeO2mixed-oxide at low temperature[J].Journal of Hazardous Materials,2012,243:117-123.
[27]ZHANG An-chao,ZHENG Wen-wen,SONG Jun,et al.Cobalt manganese oxides modified titania catalysts for oxidation of elemental mercury at low flue gas temperature[J].Chemical Engineering Journal,2014,236:29-38.
猜你喜欢
化工管理(2022年13期)2022-12-02
分子催化(2022年1期)2022-11-02
消费电子(2022年6期)2022-08-25
数学小灵通(1-2年级)(2021年10期)2021-11-05
建材发展导向(2021年16期)2021-10-12
建材发展导向(2021年12期)2021-07-22
数学小灵通(1-2年级)(2020年12期)2021-01-14
船舶标准化工程师(2020年1期)2020-06-12
智富时代(2018年3期)2018-06-11
智富时代(2018年3期)2018-06-11
