
硫化促进剂熔点测定方法分析
2015-07-31 01:48宋魁景
橡胶科技 2015年5期
宋魁景
(东北助剂化工有限公司,河北 武强 053300)
在沿用多年的旧版国家标准或行业标准中,硫化促进剂检测项目主要包括外观、加热减量、灰分、初熔点、筛余物5项。其中,初熔点是最能表征促进剂纯度的项目,其重要性不言而喻。随着科技进步和检测手段日益完善,以及满足用户要求及与国外标准接轨的需要,从2006年起国内对次磺酰胺类促进剂NOBS和CBS国家标准和行业标准进行修订更新,增加了纯度、游离胺、甲醇不溶物检测项目。之后修订更新的所有促进剂标准几乎都增加了纯度或其他附属项目,并且纯度检验多以色谱检验为仲裁。
虽然促进剂质量不仅用初熔点表征,但对于诸多橡胶助剂生产企业及用户而言,熔点测定快捷方便,加上几十年来的习惯性使用,更因熔点与纯度间的关联性,熔点测定在促进剂生产中仍占据着不可替代的重要地位。
熔点的测定方法和测定仪器多种多样,测定结果的影响因素较多。初熔点和熔点的判定至关重要,如果检测时未严谨地按规范操作,会导致测定结果重现性差且偏差较大,数据无可比性;不同实验室和检测人员产生的检测误差不同,这些因素造成无法客观、准确地进行产品质量评价。结合多年实践经验,笔者对现行促进剂标准中熔点测定方法及其测定结果的影响因素进行分析。
1 熔点测定的意义
1.1 熔点的定义
固体物质的熔点一般是指在标准大气压(1.01×105 Pa)下从固态转变成液态的温度。通常固体物质的熔点又可细分为初熔点、熔点、全熔点。物质最初熔化时的温度为初熔点,固液并存明显大面积液化时的温度为熔点,全部由固态转化为液态时的温度为全熔点或终熔点。
国内促进剂标准中要求检测的熔点均指初熔点,但许多检测仪器公司和部分国外用户习惯性使用熔点或全熔点。
结合毛细管法,硫化促进剂国家标准中将初熔点定义为当试样收缩并且在毛细管壁上开始出现液体时的温度为初熔温度。
1.2 熔点的意义
促进剂熔点之所以受到业内关注,是因为在一定范围内熔点与纯度之间存在定量关系。试样纯度越高,初熔点越高,熔程越短。如果试样中含有杂质,则初熔点降低,熔程延长。例如,当促进剂TMTD中含有1%(质量分数,下同)硫黄时,其熔点下降8 ℃;含有3%硫黄时,熔点下降21.5 ℃;含有4%硫黄时,熔点下降27.5 ℃[1]。促进剂MBTS中游离M含量超过1%时,初熔点下降较为明显,甚至低于162 ℃。可见初熔点与纯度有密切关系。因此确切掌握熔点测定方法,对准确评价促进剂质量尤为重要。
1.3 初熔点的判定
相关资料和实际检测表明,在熔点测定过程中,毛细管内试样状态变化可分为5个阶段:初始态、收缩态、初熔态、过熔态、全熔态,这5个阶段还称为变色、收缩、崩解、液化、全溶。
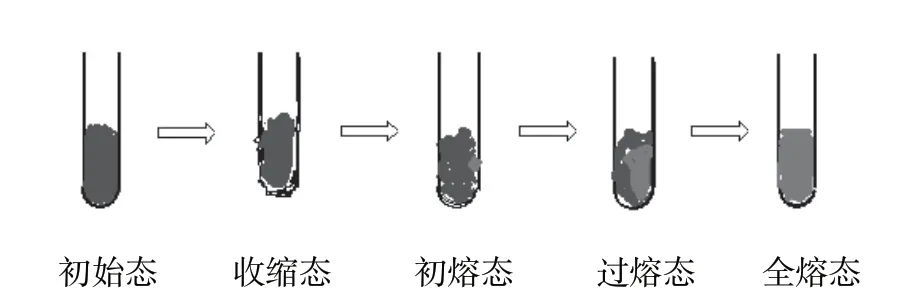
图1 毛细管内试样状态变化的5个阶段
(1)初始态:经逐渐加热,试样轻微变色,出现毛糙,试样与毛细管内壁之间出现细微雾状。如果把此时的温度视为初熔点为时过早[1]。
(2)收缩态:试样开始收缩,颜色继续加深,试样与毛细管内壁之间出现极小缝隙。
(3)初熔态:进入收缩态后,仔细观察,可以看到试样与毛细管内壁连接点出现贴壁现象,旋即潮湿并颜色加深,可观察到细小的液滴。将此时的温度定为初熔点较为确切。
(4)过熔态:随着温度继续升高,试样变色收缩明显,初溶的小液滴液化面积扩大,试样呈固液并存状态。若把此时的温度定为初熔点为时已晚,测得结果必然偏高。但也有企业检测人员习惯将此状态下的温度定为熔点。
(5)全熔态:继续升温,残存的固体完全熔化,试样完全变为液态。此时的温度为终熔点。
对于初熔点的判定,相关资料亦有详细描述:初熔之前,毛细管内的试样可能出现“发毛”“收缩”“软化”“出汗”等现象,在未出现局部液化的明显液滴和持续熔融过程时,均不作初熔判断,但如上述现象严重,过程较长或因之影响初熔点的观察时,应视为试样纯度不高的标志而予以记录。
以上是典型的熔点测定过程,但不同试样熔点测定过程中现象会有所差异。例如,有的试样可能内部先初熔,未发现明显液滴即已崩解、过熔;或未见明显收缩即已出现初熔;或者受热前期受某种微量低沸点物质气化影响,毛细管内雾化严重,难以观察,试样状态过渡不明显,影响初熔点判定,导致试样纯度不低,但熔点测定结果低等。因此必须根据不同促进剂品种及不同现象正确判定初熔点,以准确评价促进剂的质量。
2 促进剂新标准中熔点测定方法的变化
虽然修订更新后的促进剂标准已经出台数年,但仍有实验人员对其中熔点测定方法的变化细节不甚明了。2006年修订的促进剂国家标准中增加了一些附加项目,熔点测定方法结合GB/T 11409.1—1995和JISK 6220-2:2001的内容,即测定装置采用GB/T 11409.1—1995的规定,而毛细管规格、装样温度及升温速度都采用JISK 6220-2:2001的规定。之后全国橡胶与橡胶制品标准化技术委员会化学助剂分技术委员会对GB/T 11409.1—1995又进行了整合更新,形成2008版《橡胶防老剂、硫化促进剂试验方法》。目前,所有促进剂标准中初熔点测定方法均采用GB/T 11409—2008中3.1方法。
新旧版国家标准熔点测定方法的主要差异如表1所示。

表1 新旧国家标准中熔点测定方法对比
国家标准中熔点测定装置中的提勒(Thiele)管又称b形管,因其属外购加工玻璃仪器,其结构及细节要求易被忽略。实践表明,b形管结构不规范不合理有可能引入检测误差。
3 熔点测定结果的影响因素及解决措施
熔点测定结果的影响因素较多,主要归纳为以下几个方面。
(1)毛细管不符合标准要求:毛细管直径小、管壁厚度小会导致熔点测定结果偏低,反之,毛细管直径大、管壁厚度大会导致熔点测定结果偏高;毛细管底部未完全熔封,加热时空气及传热液进入会导致试样提前熔化,熔点测定结果偏低。因此,选用毛细管应严格遵守国家标准中相关要求。
(2)玛瑙研钵、毛细管、点滴板等不清洁会导致试样提前熔化,熔点测定结果偏低,严重时误差会高达数摄氏度。
(3)试样装填不实:试样必须先在玛瑙研钵内研细,然后再装样,装填紧实使热量传导均匀。密度小且易产生静电的品种如促进剂MBTS和TMTD及颗粒产品促进剂NOBS经研细过筛后装样时,更需注意试样装填的紧密程度,否则将直接影响初熔点的判定和测定结果。
(4)试样未完全干燥:试样中如果含有水分和其它溶剂,受热后水和溶剂气化使试样松动熔化,导致熔点测定结果偏低、熔程长。因此,试样必须在加热减量试验温度下烘至恒质量。
(5)升温速率不准确,加热温度未按要求匀速上升:升温速度太快会导致熔点测定结果偏高;反之,升温速度太慢会导致熔点测定结果偏低。因此,应严格按国家标准要求的升温速率(如表1所示)升温,尤其要注意的是应匀速升温,不可反复升降或中间停滞。
(6)温度计及测得值未校正:精密温度计需经过校正;如果使用全浸式温度计,必须按要求对测得值加以校正。
(7)试样毛细管及温度计水银球放置位置不规范:传热液在b形管内流动过程中,上下位置处会有温度差异,应按国家标准要求保持全浸式温度计水银球下端距液面约50 mm,局浸式温度计水银球下端达到浸没线。
(8)传热液太少,未达b形管横梁处或b形管横梁处液位偏低,不能正常循环:常温下b形管内传热液的液位应保持在横梁1/2高度以上,以确保传热液在管内快速均匀循环。
(9)初熔点的判定有误:在熔点测定的各个阶段试样的表象不同,应认真观察,准确判定初熔点,避免提前和滞后判定初熔点。
4 结语
综上所述,在熔点测定过程中,只有测定方法、检测仪器及操作规范统一,排除有可能导致熔点测定误差的因素,按要求控制升温速度,准确判定初熔点,测定结果才具有客观准确性和可比性。
猜你喜欢
四川蚕业(2022年2期)2022-11-19
橡胶科技(2022年5期)2022-07-20
玩具世界(2022年1期)2022-06-05
环境保护与循环经济(2021年7期)2021-11-02
陶瓷学报(2021年1期)2021-04-13
上海建材(2019年1期)2019-04-25
橡胶科技(2018年4期)2018-02-17
江苏农业科学(2017年10期)2017-07-21
橡胶工业(2015年11期)2015-08-01
橡胶工业(2015年3期)2015-07-29
