
EMIM离子液体离子簇模型的量子化学计算
2015-12-05 06:29戚传松
物理化学学报 2015年9期
李 巍 张 静 戚传松
(1北京石油化工学院化学工程学院, 北京 102617; 2中国科学院大学材料科学与光电技术学院, 北京 100049)
EMIM离子液体离子簇模型的量子化学计算
李 巍1,*张 静2戚传松1
(1北京石油化工学院化学工程学院, 北京 102617;2中国科学院大学材料科学与光电技术学院, 北京 100049)
以1-乙基-3-甲基咪唑(EMIM)卤化物、氟硼酸盐、三溴化物和二碘溴酸盐、氯铝酸和溴铝酸盐等不同种类EMIM离子液体为研究对象, 对多阳离子、多阴离子的离子簇模型进行了量子化学计算研究. 首先在B3LYP/6-311++G(d,p)水平上(I使用6-311G(d,p)基组)对{[EMIM]Xn}(n–1)–(X = Cl, Br, I, BF4, AlCl4, AlBr4, Br3, IBrI, FHF; n = 2, 3)和{[EMIM]2Xn'}(n'–2)–(n' = 3, 4, 5)离子簇进行构型优化, 并对卤化物和氟硼酸盐进行了振动光谱计算. 结果表明所采用理论模型在键长、键角等结构参数及红外振动光谱方面均与实验结果符合较好.同时对不同离子簇模型中阴、阳离子间相互作用能与实验熔点之间的关系进行了研究, 发现采用{[EMIM]2Xn'}(n'–2)–模型时EMIM离子液体实验熔点与阴、阳离子间相互作用能之间呈现近线性关系.
密度泛函理论; 咪唑离子液体; 相互作用能; 离子簇; 熔点
1 引 言
室温离子液体(RTILs, 简称ILs)是由不对称的有机阳离子与无机或有机阴离子构成的一类新型溶剂, 由于其环保性好等特点被认为是传统有机溶剂的有力替代者;1–4而增加离子液体的种类、充实离子液体化学的基础内容、开辟离子液体新的研究与应用领域是绿色化学的需要. 有机阳离子与阴离子之间可能的组合数目庞大, 但是能够满足特定应用需求的组合方式是有限的, 因而离子液体经常被称为“designer solvents”, 而如何设计出具有特定性质及功能的离子液体是当前的一个研究热点. 目前离子液体的设计仍然是以一种比较盲目的“tryand-error”模式进行, 并没有真正做到根据目标进行设计. 为了能够从分子水平上设计低熔点、功能化的离子液体, 以及在新型离子液体的探索中避免人力及物力的浪费, 离子液体基础性质及功能预测的理论研究势在必行.
而离子液体的理论计算研究相对于其实验研究发展较慢, 这主要是由于对于凝聚相体系至今还没有发展出一个高精度和高效性两者兼备的算法.目前常用的理论手段包括使用量子化学计算方法研究气相中离子对结构, 或是构建力场、利用分子模拟方法研究离子液体的宏观物理性质, 以及利用统计方法结合量子化学5–14或半经验方法15–19进行定量构效关系(QSPR)研究. 在过去的10年里, 基于分子描述符的QSPR研究发展迅速, 在相关的研究中多数采用半经验方法或分子力学方法优化构型, 参数的选择主要服务于精度的需要, 对预测结果影响较大的参数数量较多, 对物理化学性质的影响因素只能定性的分析; 同时, 方法应用的效果依赖于算法及个人经验, 至今为止还没有一种普适于不同种类离子液体的构效关系表达式.
在构效关系研究方面我们的目标是力图发现一种普适性强、形式简单、物理意义明确的构效关系式, 而目前我们最关注的实验性质是熔点, 这是由于低熔点是离子液体具有应用前景的首要条件; 此外He等20发现对于N-取代的甘氨酸酯化物阳离子型离子液体, 熔点与黏度呈现正相关性; Rahman等21在研究四乙铵氨基酸离子液体时则发现其离子导电性与黏度成线性相关关系. 可见, 熔点是离子液体非常重要的一项性质指标. 考虑到离子液体是一种以库仑相互作用为主的体系, 而在发生熔化过程体积发生膨胀时需要克服的阻力以阴阳离子间相互作用为主, 因此我们以量子化学计算获得的离子间相互作用为唯一的描述符, 试图建立离子液体的熔点与离子间相互作用的定量关系. 我们已经在氨基酸阳离子型离子液体、5烷基咪唑氟硼酸离子液体6和烷基咪唑卤化物7中进行了尝试并获得一定的成功. 类似的尝试也表现在部分其他小组的工作中, 但多数以失败告终, 其中Wu8和Mohajeri9等均对咪唑氨基酸离子液体的离子对进行了系统地量子化学计算研究; 而Turner10及Katsyuba小组11分别系统研究了咪唑卤化物及咪唑氟硼酸系列离子液体的离子对结构. 上述工作在咪唑离子液体研究时未能获得与实验熔点与某种分子结构性质的相关性的主要原因是由于他们均采用单个阳离子与单个阴离子组成的离子对模型, 而此模型不能真正反映凝聚相体系中真实的结构特征.6,7我们之前在研究咪唑类化合物时选择一个咪唑阳离子与2–3个阴离子组成的一价或二价分子离子为研究对象, 发现量子化学计算优化获得的结构与晶体结构实验结果更加符合, 且阴、阳离子间相互作用能与实验熔点之间存在线性关系.6,7在之前关于咪唑类离子液体的构效关系研究中, 基本是以单一种类阴离子的体系为研究对象. 在本文中, 我们在前期工作的基础上扩展研究对象, 对阴离子种类不同的常见1-乙基-3-甲基咪唑(EMIM)离子液体进行量子化学计算研究, 采用多个阳离子和多个阴离子组成的离子团簇模型,考虑不同阴离子与EMIM阳离子间的作用模式, 并进一步研究熔点与离子间相互作用能之间的构效关系.
2 研究体系及研究方法
2.1 研究体系
我们之前研究了阴离子相同的咪唑类离子液体实验熔点与离子间相互作用能之间的关系, 在本次工作中我们选择阳离子均为1-乙基-3-甲基咪唑的离子液体为研究对象, 通过检索Reaxys数据库22收集具备实验熔点的1-乙基-3-甲基咪唑化合物; 同时结合检索剑桥晶体数据库, 选择既具有实验熔点又具备晶体结构信息的常见1-乙基-3-甲基咪唑化合物作为研究对象, 具体包括: 氟硼酸盐([EMIM][BF4])、氟化物([EMIM][FHF])、氯化物([EMIM]Cl)、溴化物([EMIM]Br)、碘化物([EMIM]I)、三溴化物([EMIM][Br3])及溴铝酸盐([EMIM][AlBr4]). 此外, 氯铝酸盐([EMIM][AlCl4])、二碘溴酸盐([EMIM][IBrI])虽然未查到晶体结构信息, 但由于其阴离子部分与已知晶体结构的化合物具有相似性, 在本文工作中也进行了讨论.
2.2 理论模型的选择
通过检索剑桥晶体数据库, 得到了百余种1-乙基-3-甲基咪唑化合物的晶体结构信息. 通过观察、对比发现阴离子的尺寸、对称性对晶体中离子的排列有着至关重要的影响, 从而进一步影响离子间的相互作用模式. 本文讨论的常见EMIM离子液体中阴离子从小至大排列为(括号内数值为估算得到的离子外接球半径, 其中多原子阴离子按照晶体结构中键长数据及端原子范德华半径23估算得到): Cl–(0.18 nm)、Br–(0.20 nm)、I–(0.22 nm)、FHF–(0.25 nm)、(0.44 nm). 阴离子尺寸及对称性相近时晶体结构中阴、阳离子的排布模式也相似. 我们选择能够反映晶体结构中阴、阳离子相互作用模式的最小结构单元作为理论模型, 具体选择的离子簇如图1和图2所示,具体分析详见结果与讨论部分.
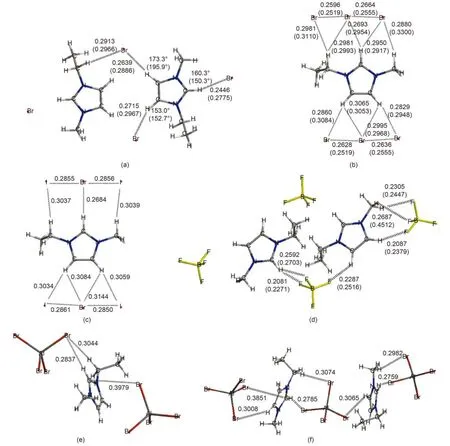
图1 EMIM离子液体离子簇优化构型Fig.1 Optimized structures of ion clusters of EMIM ILs
在我们之前的工作6,7中曾经对烷基咪唑卤化物及烷基咪唑氟硼酸盐进行过系统的量子化学计算研究, 当时采用的模型是{[EMIM]Xn}(n–1)–(n=2, 3),这一模型比较好地反映了直接与咪唑阳离子作用的周围阴离子的排布情况, 并且从该模型出发得到了离子间相互作用能与实验熔点之间的线性关系.但该模型仍然存在不足: 首先, 它并不适用于较大阴离子体系, 如[EMIM][Br3]、[EMIM][AlBr4]等情况; 此外, 对于氟硼酸体系该模型中B原子与咪唑环几乎在同一平面内, 与实际晶体结构不完全一致.在本文中我们将选取能够更好反映实际晶体结构特征的离子簇模型作为研究对象, 探究阴离子种类不同的咪唑离子液体中离子间的作用模式.
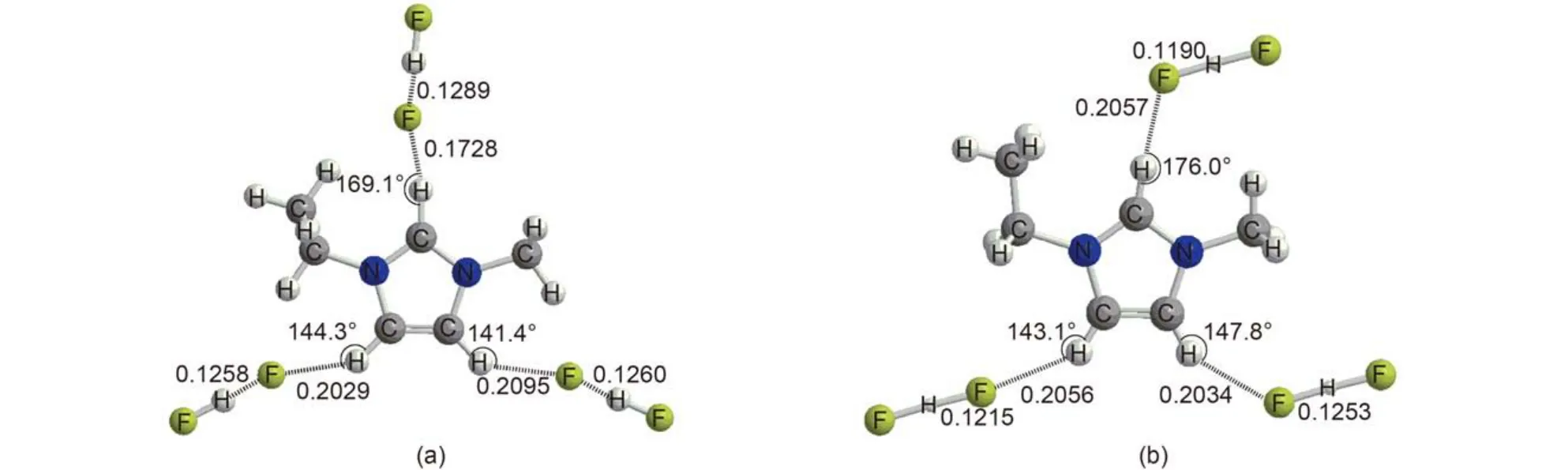
图2 {[EMIM][FHF]3}2–优化构型及晶体结构中相应结构单元Fig.2 Optimized geometry of {[EMIM][FHF]3}2–and corresponding geometry in crystal
2.3 量子化学计算
基于Gaussian 03软件包,24采用密度泛函理论(DFT)方法B3LYP和6-311++G(d,p)基组(I使用6-311G(d,p)基组), 对不同离子簇进行了构型优化并对部分离子簇优化构型进行了振动频率分析, 将得到的理论振动光谱与实验结果进行对比, 帮助指认实验红外光谱谱线的同时对不同理论模型的结果进行了对比. 此外, 利用超分子方法计算了离子簇中离子间相互作用能, 即分别代表经过结构优化的阳离子、阴离子及离子簇能量),25并且采用Counterpoise方法进行了基组重叠效应(BSSE)修正,26–28进而讨论离子间相互作用能与实验熔点之间的关系.
3 结果与讨论
3.1 优化构型及振动光谱
各离子簇的优化构型见图1及图2. 其中部分重要结构参数列于图中(图中括号部分为实验晶体结构中对应的数值, 键长量纲为nm)及相应表格中. 下面将对不同的体系进行具体分析.
3.1.1 卤化物
{[EMIM]2Br4}2–的优化构型参见图1(a), 碘化物与氯化物的优化构型和溴化物相似, 未给出具体构型图, 相应结构参数列于表1中(优化结果具有对称性, 仅列出一组数据). 如图1(a)所示的结构单元采用两种不同的取向在空间交错排布构成周期性结构.
溴化物与碘化物的优化结果均与晶体结构中的相应结构参数符合较好, 从图1(a)及表1列出的具体数值分析, Br…H距离理论结果比实验值小0.025–0.033 nm, Br…C距离均比实验值小0.010 nm; I…C的距离与实验值比较小0.005 nm左右, 这主要是由于未考虑周围离子环境而孤立考虑离子簇所造成的. 之前氯化物晶体结构的报道中C…Cl的平均键长为0.355 nm,29优化结构中C…Cl的平均值为0.346 nm, 与实验值符合较好.
为了进行对比, {[EMIM]X3}2–(X = Br, I)模型的优化构型参数7也列于表1中. 可以发现{[EMIM]2X4}2–和{[EMIM]X3}2–模型中的结构参数都比较好地接近实验晶体结构中的数据, 而{[EMIM]2X4}2–模型在键角描述方面更接近实验值, 且从描述离子间的相互作用模式来说更全面, 同时从后面3.2节的讨论中还可以看到采用{[EMIM]2X4}2–模型后其离子间相互作用能与实验熔点之间的定量关系与其他种类阴离子的EMIM化合物符合更好.
卤化物振动光谱的实验数据及不同离子簇模型的理论计算值列于表2中, 其中对于实验光谱峰的指认主要是根据与{[EMIM]2X4}2–模型理论值的比较分析来实现. 各离子簇模型的计算结果均无虚频. 由于{[EMIM]2X4}2–模型相对于其在晶体中的结构更加紧凑, 可以看到咪唑环C-H的伸缩振动频率虽然接近实验值但略高于实验值. 表2中涉及甲基和乙基的振动模式的理论计算结果明显偏离实验结果, 这主要是因为所采用模型并未考虑甲基和乙基周围环境的影响. 我们根据Scott和Radom30的处理方法, 利用{[EMIM]2X4}2–(X = Cl, Br, I)模型的甲基和乙基振动模式的计算结果, 得到校正因子0.9384.表2中括号数值即为原有计算结果乘以校正因子后的数值, 经校正后与实验结果符合较好. 而其他振动模式, 如咪唑环上的C-H伸缩振动等, 是在考虑了周围离子环境条件下计算得到的, 因此未作进一步校正. [EMIM]Cl离子对的最稳构型及{[EMIM]Cl3}2–模型的振动光谱计算结果也列于表2中, 其振动模式与{[EMIM]2X4}2–模型不同的直接列于相应数值下方. 通过对比不难发现各模型在咪唑环甲基和乙基的振动情况描述方面差别不大; 但在咪唑环C―H的伸缩振动描述上本研究中所采用的模型与实验值31符合更好, 而离子对模型的结果明显偏离实验值.
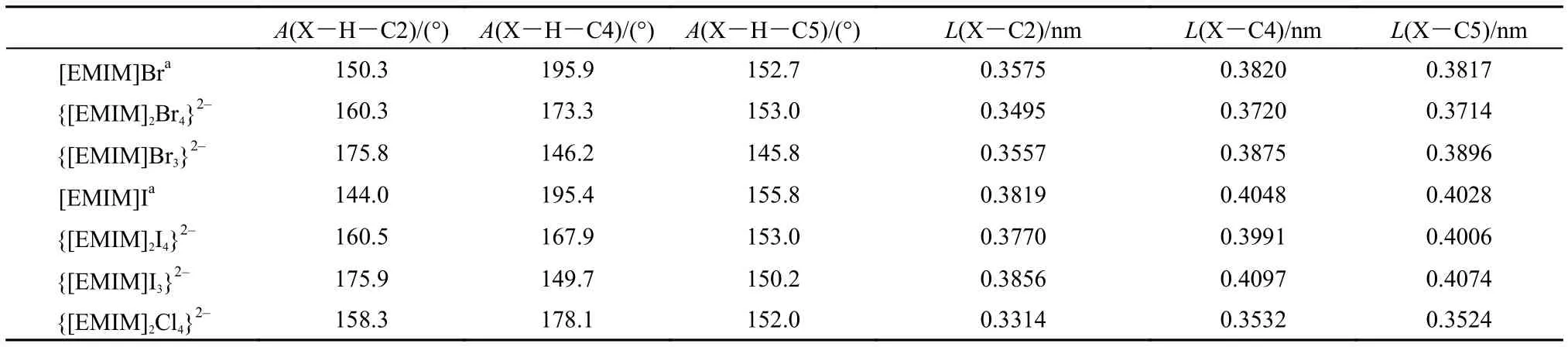
表1 EMIM卤化物晶体结构及各优化构型结构参数Table1 Structural parameters in crystal and optimized ion clusters of EMIM halides
3.1.2 氟硼酸盐
与卤化物情况相似, 在EMIM氟硼酸盐晶体结构中, 如图1(d)所示的结构单元采用两种不同取向在空间交错排布形成周期性结构. {[EMIM]2[BF4]4}2–优化构型与实验晶体结构中的相应单元比较, 咪唑环的排布相对松散: 晶体结构中咪唑环中心连线与咪唑环夹角大于{[EMIM]2[BF4]4}2–优化构型中的相应夹角(均取锐角); 咪唑环之间垂直距离实验值比理论模型值短约0.01 nm. 在我们之前工作中采用的{[EMIM][BF4]3}2–模型中阴离子的中心B原子与咪唑环几乎在同一平面, 而{[EMIM]2[BF4]4}2–优化构型中与咪唑环C2原子相近的阴离子中心B原子与EMIM咪唑环不同面, 后者更加符合晶体结构中的离子排布特征.
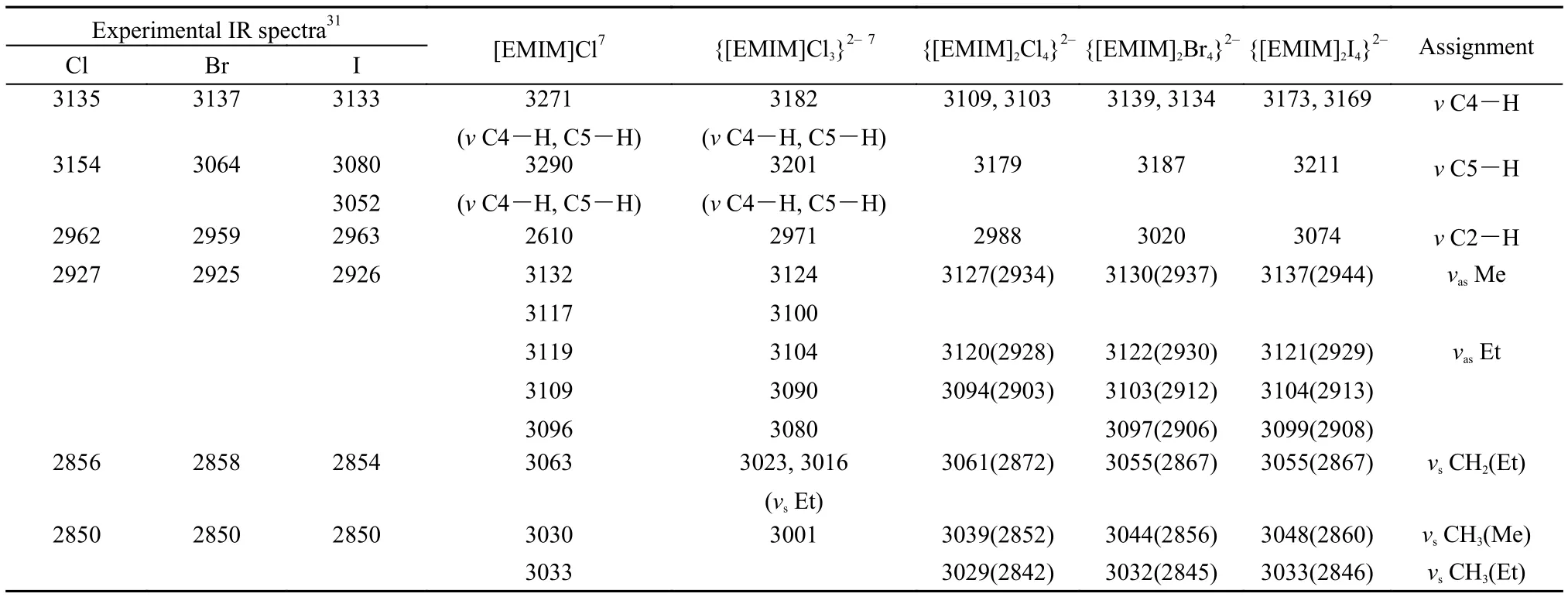
表2 [EMIM]X红外光谱实验及理论计算值(单位cm–1)Table2 Experimental and calculated IR spectrum data (unit in cm–1) of [EMIM]X
对EMIM氟硼酸盐的簇模型也进行了振动频率计算, 虽然{[EMIM]2[BF4]4}2–的计算结果中存在虚频, 但考虑到该模型的初始构型是从EMIM氟硼酸晶体结构中的相应单元出发, 因此没有考虑其他可能的构型. EMIM氟硼酸盐的红外光谱实验值11及不同理论模型计算值列于表3中. Katsyuba等11对[EMIM][BF4]离子对模型进行了细致的研究, 但无论哪种离子对的稳定构型其红外光谱中缺失对应于实验值1619 cm–1的振动模式, 在我们的离子簇模型中指认该振动模式为咪唑环C2-N3与C4-C5的反对称协同振动; 此外, 该文献中1576 cm–1被指认为C=C振动, 而我们的计算结果中其对应于N1-C2-N3反对称伸缩. 由于所研究离子簇模型未考虑周围其他离子的影响, 得到的阴、阳离子间距离小于实验值, 咪唑环C-H振动频率均高于实验值. 咪唑甲基和乙基的相应振动频率采用卤化物中得到的修正因子0.9384进行修正(表3中括号中数值),修正后结果与实验值符合较好.
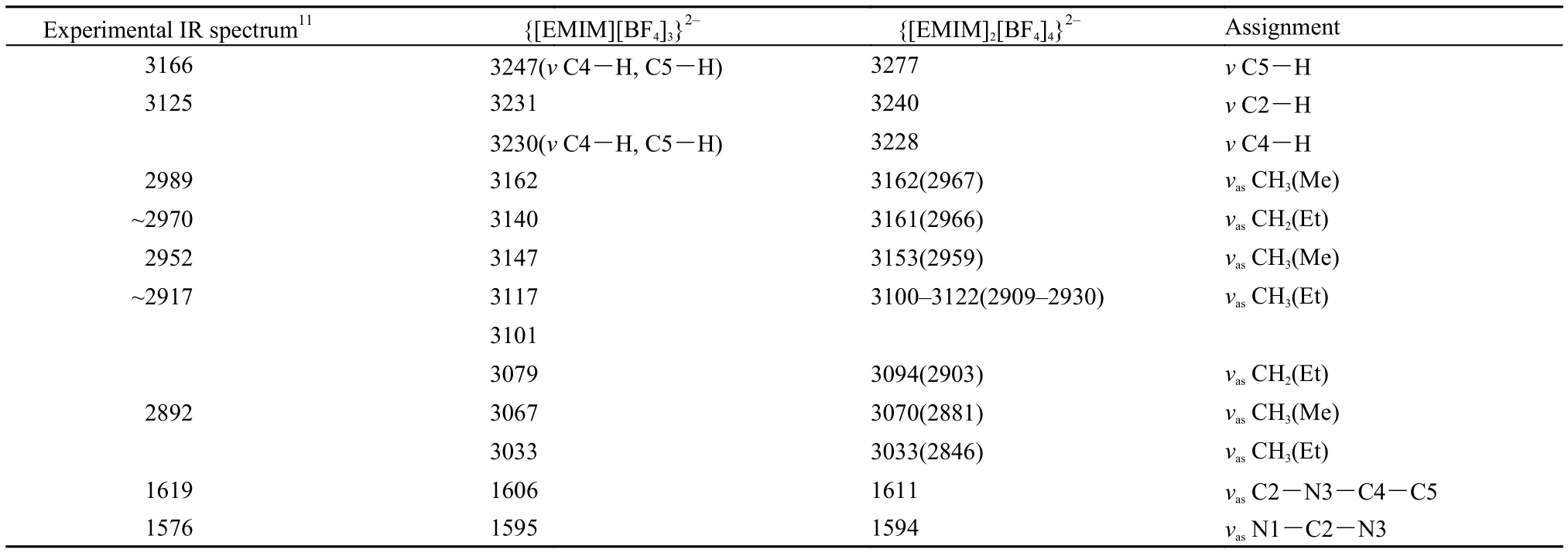
表3 [EMIM][BF4]红外光谱实验及理论计算值Table3 Experimental and calculated IR spectrum data of [EMIM][BF4]
3.1.3 氟化物
对于线性阴离子, 如FHF–和离子, 其与EMIM形成的化合物晶体结构呈层状排布; 但由于二者尺寸相差较大, 每层中离子的排布方式并不相同. 在[EMIM][FHF]的晶体结构中, 每一层中每个阳离子直接与3个阴离子相互作用, 而每个阴离子直接与3个阳离子直接发生相互作用. {[EMIM][FHF]3}2–的优化构型和晶体结构中的重复单元结构如图2所示. 从图中可以看出由于空间排布的需要, 位于C2及C4附近的阴离子取向相对于气相中{[EMIM][FHF]3}2–单独存在时发生改变.
3.1.4 三溴化物及二碘溴酸盐
[EMIM][Br3]晶体结构中沿c轴方向咪唑阳离子与交错呈列状排布, 根据这一特点选择如图1(b)所示的{[EMIM][Br3]2}–离子簇模型并进行优化.由图中数据可见优化构型与相应晶体结构中对应部分的差别主要在于离子簇中咪唑环上方略偏向乙基一侧, 下方略偏离直线结构(键角为173.8°), 同时Br3–在所研究离子簇中更加松散. 类似地对{[EMIM][IBrI]2}–进行结构优化, 结果列于图1(c). 同时为了建立离子间相互作用能与实验熔点之间的定量关系, 对呈列状排布的{[EMIM]2[Br3]3}–和{[EMIM]2[IBrI]3}–模型也进行了优化和能量计算.
3.1.5 氯铝酸盐及溴铝酸盐
对于EMIM溴铝酸盐, 阴、阳离子尺寸相当, 晶体结构中离子的空间排布更加规律, 阴、阳离子沿a轴及c轴均呈列状排布, 且阴、阳离子列呈锯齿状交替排布(参见图1(f)). 本文中研究的离子簇模型描述阴、阳离子交替排布的情况.
{[EMIM][AlBr4]2}–优化构型见图1(e), 具体结构参数列于表4(对阴离子部分相同原子进行编号时按照从左至右自小而大的方式), 优化结果基本能够反映出晶体结构中离子间的作用模式. 相对于前面的离子簇模型, 此模型的优化构型参数偏离实验数据较大, 最明显的偏离是两个阴离子的中心Al所成直线与咪唑环平面之间的夹角(锐角)比晶体结构中的相应数值小, 因而表4中二面角及键角数据与实验值偏离较大, 相应的键长偏离实验数据~0.07 nm. 当阴离子尺寸变大, 离子堆叠情况更加复杂时, 周围离子的影响越来越不可忽略. 同时对{[EMIM]2[AlX4]3}–(X = Cl, Br)进行了构型优化, {[EMIM]2[AlBr4]3}–的优化结果参见图1(f).
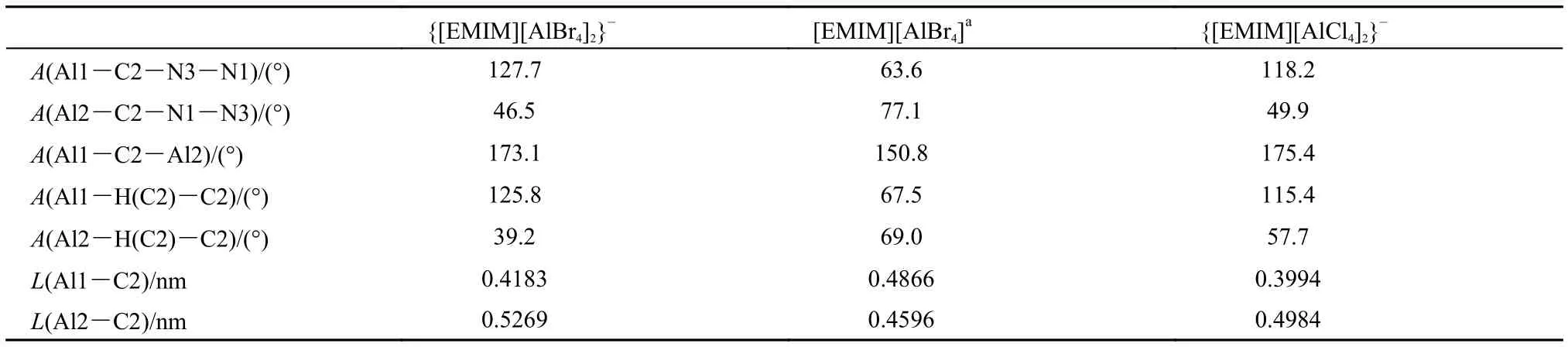
表4 溴铝酸盐晶体结构及{[EMIM][AlBr4]2}–, {[EMIM][AlCl4]2}–优化构型结构参数Table4 Structural parameters in [EMIM][AlBr4] crystal and optimized ion clusters of {[EMIM][AlBr4]2}–and {[EMIM][AlCl4]2}–
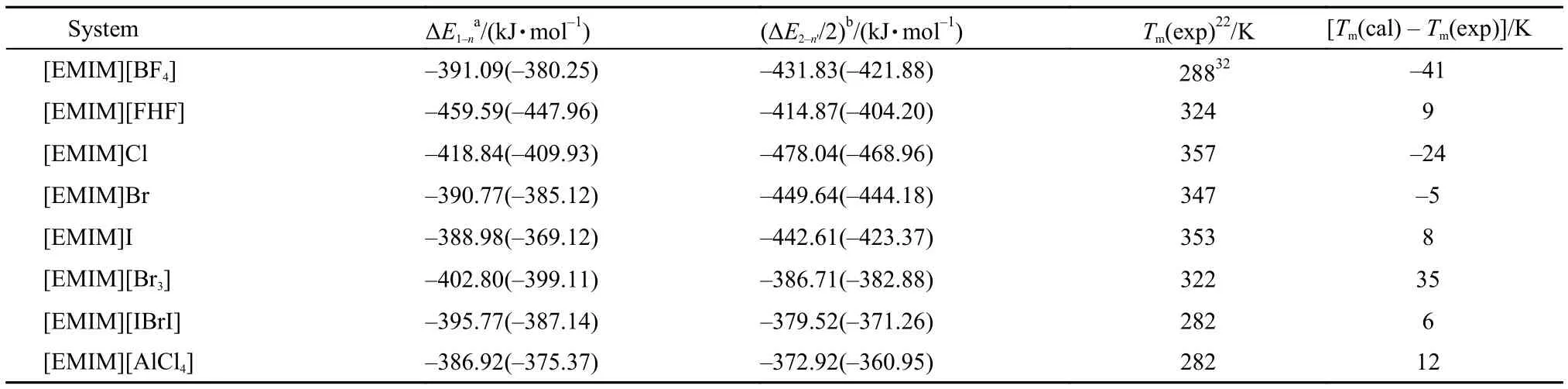
表5 离子簇模型中离子间相互作用能及实验熔点Table5 Ion interaction energy and experimental melting points of ion cluster model
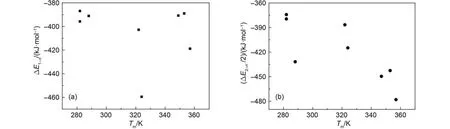
图3 EMIM离子液体离子间相互作用能与实验熔点之间的关系Fig.3 Relationships between the ion interaction energies and the experimental melting points of EMIM ion liquids
3.2 离子间相互作用能与实验熔点之间的关系
所研究体系的{[EMIM]Xn}(n–1)–(X = Cl, Br, I, BF4, AlCl4, Br3, IBrI, FHF; n = 2, 3)和{[EMIM]2Xn'}(n'–2)–(n' = 3, 4, 5)模型的离子间相互作用能列于表5中(括号中数据为经过BSSE校正后的结果). 其中, ΔE1–n、ΔE2–n'分别代表{[EMIM]Xn}(n–1)–和{[EMIM]2Xn'}(n'–2)–模型的阴、阳离子间相互作用能. 从表5中数据可以看到除[EMIM]I体系外BSSE修正数值均较小. 不同体系的实验熔点(主要来自文献,22,32当有不同的熔点被报道时采用平均值)也列于表5中.
图3更直观地反映了实验熔点与各离子簇模型离子间相互作用能ΔE1–n、ΔE2–n'/2 之间的关系. 可以看到对于阴离子不同的EMIM离子液体, 采用{[EMIM]2Xn'}(n'–2)–模型时实验熔点与阴、阳离子间相互作用能之间趋向线性关系. 根据8个数据点线性拟合得到方程: Tm= –125.40 – 0.92091 × ΔE, 决定系数|R| = 0.74. 采用上述拟合方程预测得到的熔点值Tm(cal)与实验值Tm(exp)的差值也列于表5中. 结果表明采用能够反映晶体结构特征的多阳离子、多阴离子的离子簇模型, 阴、阳离子间相互作用能与实验熔点存在定量关系.
4 结 论
对1-乙基-3-甲基咪唑卤化物、氟硼酸盐、三溴化物和二碘溴酸盐、氯铝酸和溴铝酸盐进行了离子簇模型研究, 所涉及的EMIM离子液体涵盖了不同种类、不同尺寸阴离子的体系. 我们首先在B3LYP/6-311++G(d,p)水平上(对I使用6-311G(d,p)基组)分别对{[EMIM]2X4}2–(X = Cl, Br, I, BF4)、(X' = AlCl4, AlBr4, Br3, IBrI)、{[EMIM][FHF]3}2–、{[EMIM]2[FHF]5}3–共14种离子簇进行结构优化, 并对卤化物和氟硼酸盐进行了振动光谱计算. 结果表明对于所选定的体系, 多阳离子、多阴离子的离子簇模型能够更好地反映相应晶体中的结构及阴、阳离子相互作用特征. 我们同时对阴、阳离子间相互作用能与实验熔点之间的关系进行了研究, 发现采用2个咪唑阳离子和n个阴离子组成的模型时不同类型的EMIM离子液体晶体实验熔点与阴、阳离子间相互作用能之间呈现近线性关系. 借助理论计算预言离子液体性质一直是人们的一个研究目标, 我们在之前离子液体熔点构效关系研究中发现, 若想获得普适性强、形式简单、物理意义明确的定量构效关系表达式需要以合理的结构模型为出发点. 本文的工作进一步证明了对于离子液体这种特殊的体系, 有可能利用离子簇模型模拟晶体中的结构特征, 并进一步通过离子间相互作用能预言实验熔点. 我们希望通过我们的工作为离子液体的理性化设计提供有力的理论支持.
(1)Rogers, R. D.; Seddon, K. R. Science 2003, 302, 792. doi: 10.1126/science.1090313
(2)Li, R. X. Green Solvent—the Synthesis and Application of Ionic Liquids; Chemical In dustry Press: Beijing, 2004. [李汝雄. 绿色溶剂—离子液体的合成与应用. 北京: 化学工业出版社, 2004.]
(3)Zhang, S. J.; Lü, X. M. Ionic Liquids—from Fundamentals to Applications; Scientific Publish Ltd.: Beijing, 2006. [张锁江, 吕兴梅. 离子液体—从基础研究到工业应用. 北京: 科学出版社, 2006.]
(4)Hallet, J. P.; Welton, T. Chem. Rev. 2011, 111, 3508. doi: 10.1021/cr1003248
(5)Li, W.; Wu, X. M.; Qi, C. S.; Rong, H.; Gong, L. F. J. Mol. Struct.: Theochem 2010, 942, 19. doi: 10.1016/j.theochem. 2009.11.027
(6)Li, W.; Qi, C. S.; Wu, X. M.; Rong, H.; Gong, L. F. Acta Phys. -Chim. Sin. 2011, 27, 2059. [李 巍, 戚传松, 吴新民,荣 华, 龚良发. 物理化学学报, 2011, 27, 2059.] doi: 10.3866/PKU. WHXB20110914
(7)Li, W.; Qi, C. S.; Rong, H.; Wu, X. M.; Gong, L. F. Chem. Phys. Lett. 2012, 542, 26. doi: 10.1016/j.cplett.2012.05.072
(8)Wu, Y.; Zhang, T. T. J. Phys. Chem. A 2009, 113, 12995. doi: 10.1021/jp906465h
(9)Mohajeri, A.; Ashrafi, A. J. Phys. Chem. A 2011, 115, 6589. doi: 10.1021/jp1093965
(10)Turner, E. A.; Pye, C. C.; Singer, R. D. J. Phys. Chem. A 2003, 107, 2277. doi: 10.1021/jp021694w
(11)Katsyuba, S. A.; Zvereva, E. E.; Vidis, A.; Dyson, P. J. J. Phys. Chem. A 2007, 111, 352. doi: 10.1021/jp064610i
(12)Xiao, X.; Guo, M.; Pei, Y.; Zheng, Y. Spectrochimica Acta Part A 2011, 78, 1492.
(13)Rao, S. S.; Gejji, S. P. Computational and Theoretical Chemistry 2015, 1057, 24. doi: 10.1016/j.comptc.2015.01.012
(14)García, G.; Atilhan, M.; Aparicio, S. Chem. Phys. Lett. 2014, 610–611, 267.
(15)Katritzky, A. R.; Lomaka, A.; Petrukhin, R.; Jain, R.; Karelson, M.; Visser, A. E.; Rogers, R. D. J. Chem. Inf. Comput. Sci. 2002, 42, 71. doi: 10.1021/ci0100503
(16)Zhang, S. J.; Sun, N.; He, X. Z.; Lu, X. M.; Zhang, X. P. J. Phys. Chem. Ref. Data 2006, 35, 1475. doi: 10.1063/1.2204959
(17)Varnek, A.; Kireeva, N.; Tetko, I. V.; Baskin, I. I.; Solov’ev, V. P. J. Chem. Inf. Model. 2007, 47, 1111. doi: 10.1021/ci600493x
(18)Ren, Y. Y.; Qin, J.; Liu, H. X.; Yao, X. J.; Liu, M. C. QSAR Comb. Sci. 2009, 28, 1237. doi: 10.1002/qsar.v28:11/12
(19)Kowsari, M. H.; Alavi, S.; Najafi, B.; Gholizadeh, K.; Dehghanpisheh, E.; Ranjbar, F. Phys. Chem. Chem. Phys. 2011, 13, 8826. doi: 10.1039/c0cp02581j
(20)He, L.; Tao, G. H.; Parrish, D. A.; Shreeve, J. M. J. Phys. Chem. B 2009, 113, 15162. doi: 10.1021/jp905079e
(21)Rahman, M. B. A.; Jumbri, K.; Basri, M.; Abdulmalek, E.; Sirat, K.; Salleh, A. B. Molecules 2010, 15, 2388. doi: 10.3390/ molecules15042388
(22)The Reaxys Database, http://www.reaxys.com.
(23)Hu, S. Z.; Zhou, Z. H.; Cai, Q. R. Acta Phys. -Chim. Sin. 2003, 19, 1073. [胡盛志, 周朝晖, 蔡启瑞. 物理化学学报, 2003, 19, 1073.] doi: 10.3866/PKU.WHXB20031118
(24)Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 03, Revision E.01; Gaussian Inc.: Wallingford, CT, 2004.
(25)Morrow, T. I.; Maginn, E. J. J. Phys. Chem. B 2002, 106, 12807. doi: 10.1021/jp0267003
(26)van Duijneveldt, F. B.; van Duijneveldt-van de Rijdt, J. G. C. M.; van Lenthe, J. H. Chem. Rev. 1994, 94, 1873. doi: 10.1021/ cr00031a007
(27)Jansen, H. B.; Ros, P. Chem. Phys. Lett. 1969, 3, 140. doi: 10.1016/0009-2614(69)80118-1
(28)Boys, S. F.; Bernardi, F. Mol. Phys. 1970, 19, 553. doi: 10.1080/00268977000101561
(29)Dymek, C. J.; Grossie, D. A.; Fratini, A. V. J. Mol. Struct. 1989, 213, 25. doi: 10.1016/0022-2860(89)85103-8
(30)Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502. doi: 10.1021/jp960976r
(31)Elaiwi, A.; Hitchcock, P. B.; Seddon, K. R.; Srinivasan, N.; Tan, Y. M.; Welton, T.; Zora, J. A. J. Chem. Soc. Dalton Trans. 1995, 3467.
(32)Wang, J.; Yang, X. Z.; Wu, S. D.; Li, G. S. Properties and Applications of Ionic Liquids; China Textile &Apparel Press: Beijing, 2006. [王 军, 杨许召, 吴诗德, 李刚森. 离子液体的性能及应用. 北京: 中国纺织出版社, 2006.]
Quantum Chemistry Calculations of Ion Cluster Models of EMIM Ionic Liquids
LI Wei1,*ZHANG Jing2QI Chuan-Song1
(1College of Chemical Engineering, Beijing Institute of Petro-Chemical Technology, Beijing 102617, P. R. China;2College of Materials Science and Optoelectronics Technology, University of Chinese Academy of Sciences, Beijing 100049, P. R. China)
Different types of 1-ethyl-3-methylimidazolium (EMIM) ionic liquid compounds, including halides, tetrafluoroborate, tribromide, diiodobromate, chloroaluminate, and bromine aluminate, have been investigated using quantum chemical calculations. First, geometry optimizations of the ion systems, including {[EMIM]Xn}(n–1)–(X = Cl, Br, I, BF4, AlCl4, AlBr4, Br3, IBrI, FHF; n = 2, 3) and {[EMIM]2Xn'}(n’–2)–(n' = 3, 4, 5), were performed using the density functional theory (DFT) B3LYP method together with the 6-311++G(d,p) (6-311G(d,p) for I) basis set. The vibrational spectra were also calculated for the EMIM halides and tetrafluoroborate. The obtained structures and vibrational spectra were consistent with experimental results. In addition, a linear correlation between melting point and interaction energy was obtained for the {[EMIM]2Xn'}(n'–2)–models of the compounds studied.
Density functional theory; Imidazolium ionic liquid; Interaction energy; Ion cluster; Melting point
O641
10.3866/PKU.WHXB201507071
Received: March 30, 2015; Revised: July 6, 2015; Published on Web: July 7, 2015.
*Corresponding author. Email: liwei77@bipt.edu.cn; Tel: +86-10-81292127.
The project was supported by the Breeding Project of Outstanding Academic Leaders of Beijing Institute of Petro-Chemical Technology, China (BIPT-BPOAL-2014).
北京石油化工学院优秀学科带头人培育计划(BIPT-BPOAL-2014)资助项目
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
陶瓷学报(2021年1期)2021-04-13
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
上海建材(2019年1期)2019-04-25
衡阳师范学院学报(2016年3期)2016-07-10
橡胶工业(2015年5期)2015-08-29
现代农业(2015年3期)2015-02-28
应用化工(2014年1期)2014-08-16
火炸药学报(2014年3期)2014-03-20
郑州大学学报(理学版)(2013年2期)2013-03-11
郑州大学学报(理学版)(2012年4期)2012-03-25
