
褐煤表面吸附水分子的微观机理
2016-05-06 02:19高正阳吕少昆李晋达杨朋飞陈传敏
动力工程学报 2016年4期
高正阳, 吕少昆, 李晋达, 杨朋飞, 陈传敏
(1. 华北电力大学 能源动力与机械工程学院, 河北保定 071003;
2. 广东粤电靖海发电有限公司, 广东揭阳 515223;
3. 华北电力大学 环境科学与工程学院, 河北保定 071003)
褐煤表面吸附水分子的微观机理
高正阳1,吕少昆1,李晋达2,杨朋飞1,陈传敏3
(1. 华北电力大学 能源动力与机械工程学院, 河北保定 071003;
2. 广东粤电靖海发电有限公司, 广东揭阳 515223;
3. 华北电力大学 环境科学与工程学院, 河北保定 071003)
摘要:应用密度泛函理论在分子水平上研究了水在褐煤表面的微观吸附机理,在B3LYP/6-311G+(d,p)优化基础上,采用Gaussian09软件程序包计算得到褐煤表面构型及其不同煤水吸附构型,采用完全均衡校正法对其相互作用能校正重叠误差.运用Multiwfn程序和VMD软件,应用静电势分布图、散点图和约化密度梯度(RDG)填色等值图对不同褐煤表面模型进行图形化分析.结果表明:褐煤表面对水分子的吸附为物理吸附,属于弱相互作用,这些弱相互作用力以氢键作用为主,其余为范德华弱相互作用;羟基、羧基对水的吸附性最强,其他含氧基团(如醚键、甲氧基等)次之,苯环最弱;水分子吸附形成的相互作用区域并不简单地集中于某个基团或某个原子,而是与褐煤局部区域都形成一定的相互作用,这不仅增强了水分子的吸附作用,而且对褐煤结构产生较大影响.
关键词:褐煤表面; 水分子; 氢键; 密度泛函理论; RDG填色等值图
褐煤是一种高含水量的低阶煤种[1],其煤化程度较低.褐煤的微孔结构非常复杂,且褐煤分子拥有多支链结构,支链上分布有多种氧官能团[2],主要有羧基、羟基、羰基和醚键等[3].褐煤含氧官能团的数量是影响褐煤含水量的主要因素,含氧官能团化学活性较高,作为质子传递体性质的水分子多结合在褐煤官能团附近,其中羧基和羟基活性最高,对水的吸附性最强[4-5].
目前有关煤水吸附的研究主要是基于实验[6],无法从微观上解释褐煤与水分子的结合机理.量子化学是一种可靠的微观机理研究方法,采用该方法对煤进行了一些研究,主要包括煤的结构[7]、煤的热化学性质[8]、煤表面、CO2与CH4等气体的相互作用[9]以及煤的热解行为和机理[10].然而针对褐煤的量子化学研究多采用带有单一官能团的一两个苯环的简化褐煤结构,对褐煤性质的表现存在不足.笔者应用量子化学密度泛函理论构建的褐煤模型是由环戊烯链接多个含多种官能团的苯环组成,更全面地展示了褐煤的结构和性质,在此模型基础上研究褐煤与水之间的相互作用.从微观上看,水在褐煤表面的吸附是物理吸附,其本质是褐煤表面与水分子间的弱相互作用力.笔者应用量子化学方法首先计算褐煤表面静电势,对苯环、环戊烯和各个官能团附近区域的静电势进行研究,从静电势方面对煤水相互作用进行分析;然后计算不同煤水吸附构型的吸附作用能,对吸附作用能层次进行归纳;最后图形化展示了褐煤表面与水分子弱相互作用的强度、位置和类型,对氢键作用和范德华相互作用的作用方式和作用区域进行了详细解释.
1计算方法
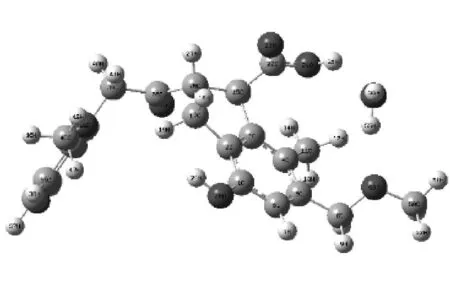
针对褐煤结构的化学特点,在一组得到广泛认可和使用的、表示不同煤阶煤结构的化学结构模型[11]的基础上,结合Wender模型[12]与Benzie等[13]提出的褐煤基本结构,采用截断和加氢饱和或甲基饱和的方法适度简化,应用量子化学Gaussian09软件程序包,采用密度泛函理论(DFT),在B3LYP/6-311G+(d,p)水平上计算得到了褐煤表面的几何优化构型,如图1所示.由图1可知,褐煤表面模型含有多种含氧官能团、多个苯环和环戊烯结构.
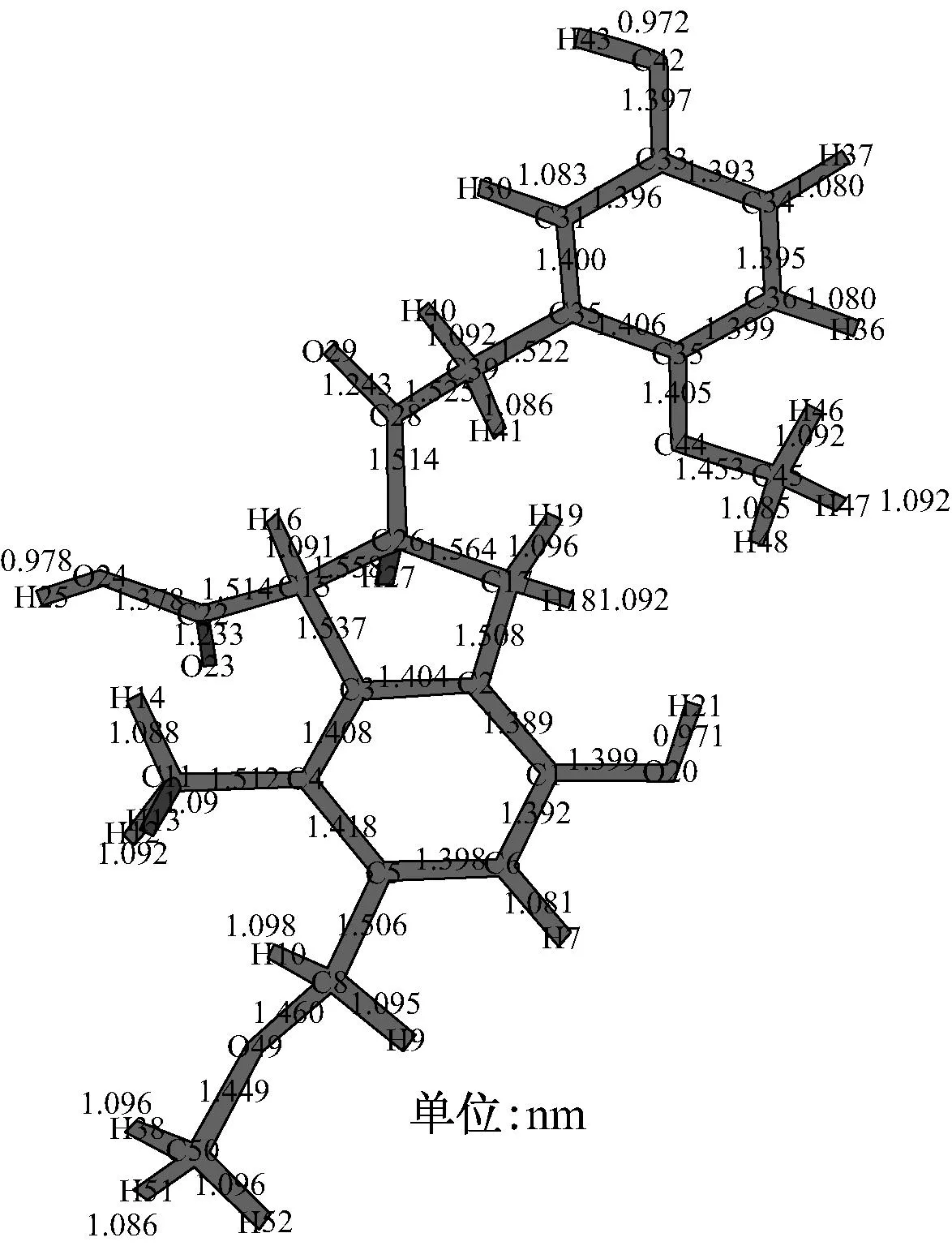
图1 褐煤表面优化结构及其键长
在图1中褐煤表面模型上,通过Multiwfn软件[14]的定量分子表面分析功能结合VMD软件[15]计算得到褐煤表面静电势分布图(见图2).考虑到褐煤表面的静电势分布情况,选取12个不同的水分子着附位置,在B3LYP/6-311G+(d,p)水平下模拟水分子在褐煤表面上的吸附过程,从而得到12种不同的煤水吸附复合物以及不同复合物下的键长和相互作用能,并采用约化密度梯度(RDG)填色等值图分析氢键的强度和位置,研究褐煤中不同含氧官能团与水分子的相互作用.本文涉及的分子表面均为电子密度ρ=0.001e/r3的等值面,其中e为电子数,r为玻尔半径.
2结果及分析
2.1褐煤表面静电势分析
化学反应的进行首先需要分子间的相互吸附、接近,在研究煤的气化、自燃和干燥等课题中,气体分子被吸附到煤表面的过程更是其研究的最重要步骤.根据文献[16]中的结论,褐煤对水的吸附主要依据分子间作用力和氢键作用.而本文研究的褐煤吸附的第一层水分子以短程相互作用力为主,主要表现为氢键作用,故分析褐煤表面静电势分布对解释水分子吸附到褐煤表面的着附位置和结合模式有重要意义.
褐煤表面静电势分布如图2(a)所示,不同静电势区间褐煤分子表面积分布如图2(b)所示.从图2可以看出,褐煤表面区域中占褐煤表面不到15%的表面区域表现出较大的负静电势(<-104.5 kJ/mol),且多为含氧官能团中氧原子对应的区域,如羟基(—OH)、羧基(—COOH)中的氧原子.含氧官能团(羟基、羧基)中的氢原子对应的表面区域则表现出较大的正静电势(>+104.5 kJ/mol),且整个褐煤表面较强的正静电势区域基本集中在这些氢原子周围.静电势绝对值较小的表面区域(-62.7~+62.7 kJ/mol)占据70%左右,多为苯环和甲基等所在区域.褐煤表面静电势的最大值和最小值分别出现在羟基的氢原子和氧原子周围区域上,对应数值分别为+209.67 kJ/mol和-184.630 6 kJ/mol.从图2还可以看出,褐煤基团的另一羟基和羧基中的氢原子和氧原子周围区域也出现了静电势的极大值和极小值,在数值上仅稍小于其最大值和最小值.
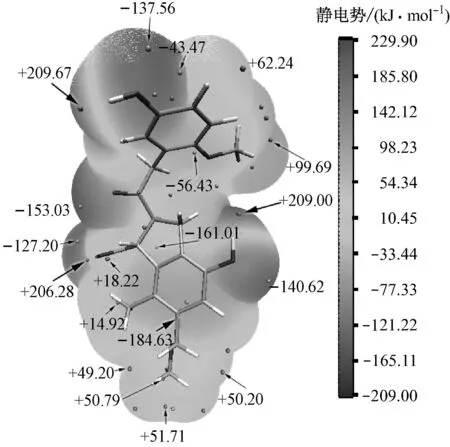
(a)

(b)
分析褐煤表面静电势可知,羟基和羧基等含氧官能团中的氢原子周围区域表现出极大的正静电势,氧原子周围区域表现出极大的负静电势,对以静电力为主要吸附力的第一层水分子具有很强的吸附性,因此H2O与褐煤中的含氧官能团之间形成的是氢键作用,且褐煤中的这些含氧官能团往往能先吸附到H2O而形成稳定的氢键复合物.当然,褐煤表面模型中的其他区域也具有一定的极大或极小静电势,H2O也与这些区域之间存在一定的弱相互作用.
2.2几何构型和相互作用能
图3给出了H2O在褐煤表面模型不同基团形成的煤水吸附构型.由图3可知,H2O在被吸附时能够在褐煤表面定向.H2O中的氢原子靠近褐煤表面的负静电势区域,氧原子靠近褐煤表面的正静电势区域.表1给出了褐煤表面吸附H2O而形成的12种复合物未校正和校正后的相互作用能和几何参数,其中ΔE为相互作用能,ΔE′为考虑基组重叠误差、能量校正后的相互作用能,D为二面角.从表1可以看出,12种复合物未校正和校正后的相互作用能均为负值且数值不大,说明褐煤表面吸附H2O形成的复合物为稳定构型,且属于物理吸附范畴.复合物12相互作用能的绝对值最大,为-49.792 16 kJ/mol,说明相比其他11种复合物,H2O与褐煤表面模型所形成的平衡吸附构型最为稳定.对比相互作用能的绝对值,还可以发现H2O吸附在褐煤表面模型中羟基和羧基上的相互作用能远大于其吸附在其他活性基团上的相互作用能.而H2O吸附在褐煤表面模型上的苯环形成的复合物的相互作用能绝对值最小,仅为-9.85 kJ/mol,这也说明H2O更容易被褐煤上的羟基和羧基所吸附,而不容易被苯环吸附.
相互作用能的大小揭示了H2O与褐煤表面的结合程度,为了能进一步揭示H2O着附在褐煤表面模型对褐煤表面结构的影响,选取了该褐煤表面模型中桥氧键两端苯环所在平面的二面角D(即C31、C33、C2、C3)作为褐煤表面模型结构扭曲变形程度的参考数值.

复合物1
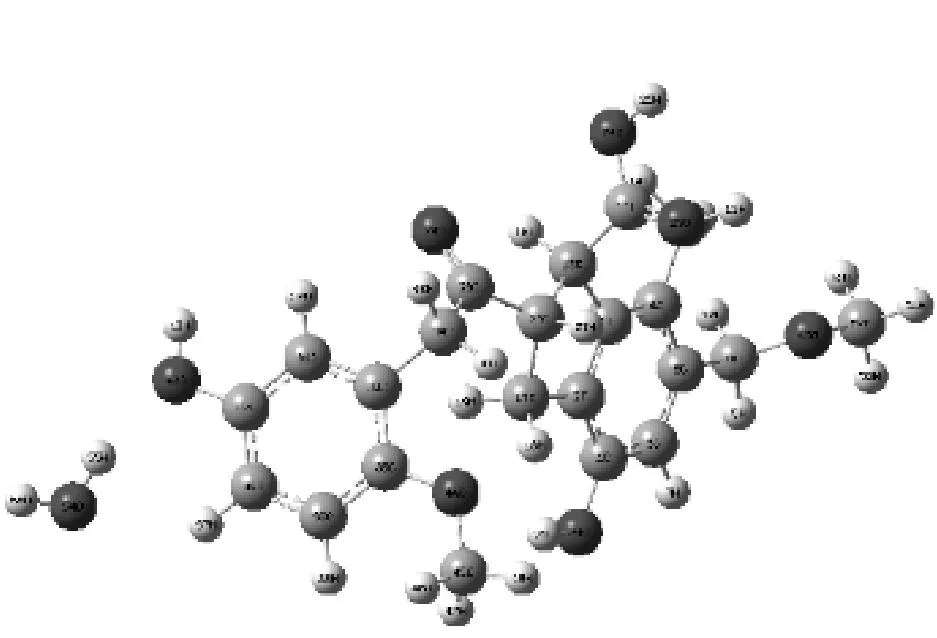
复合物2

复合物3

复合物4
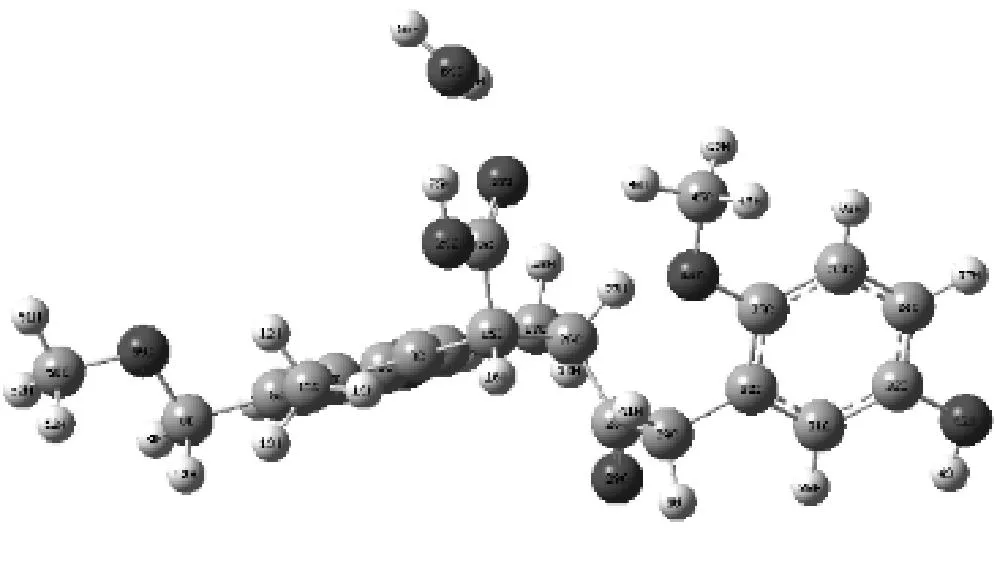
复合物5

复合物6

复合物7
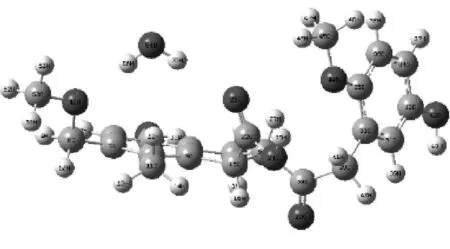
复合物8
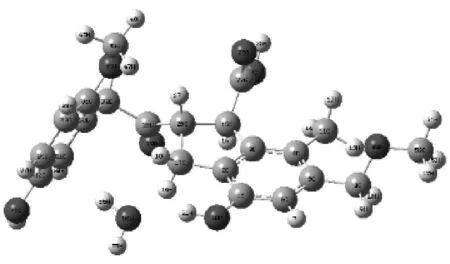
复合物9
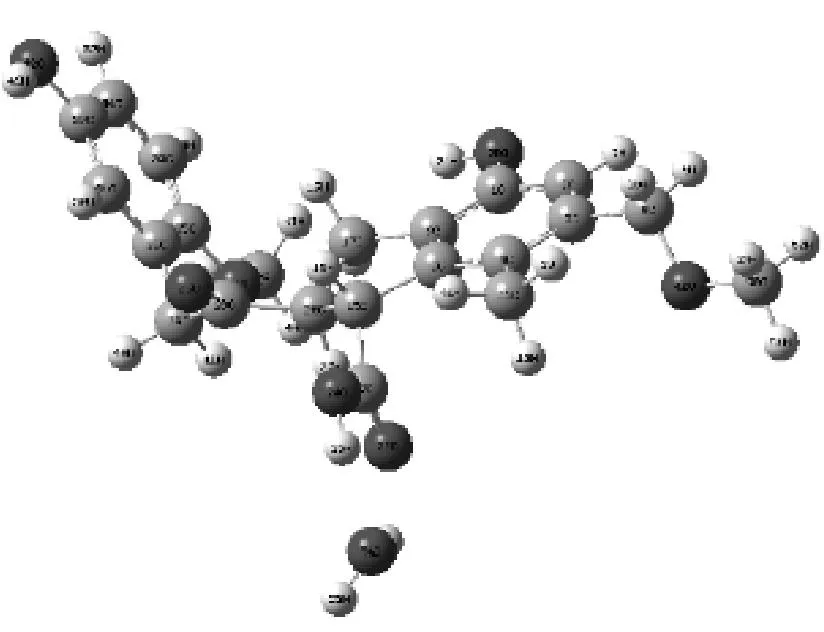
复合物10
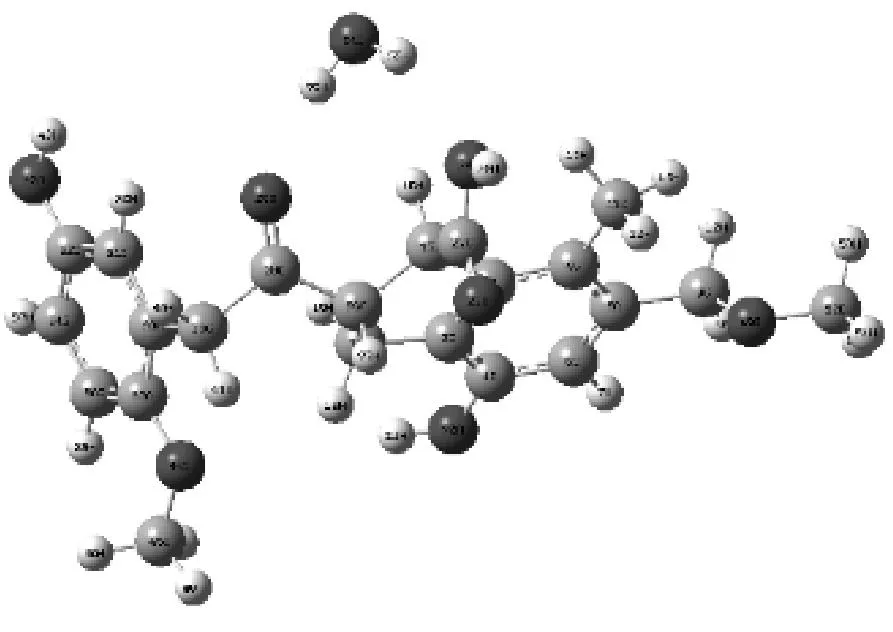
复合物11
复合物12
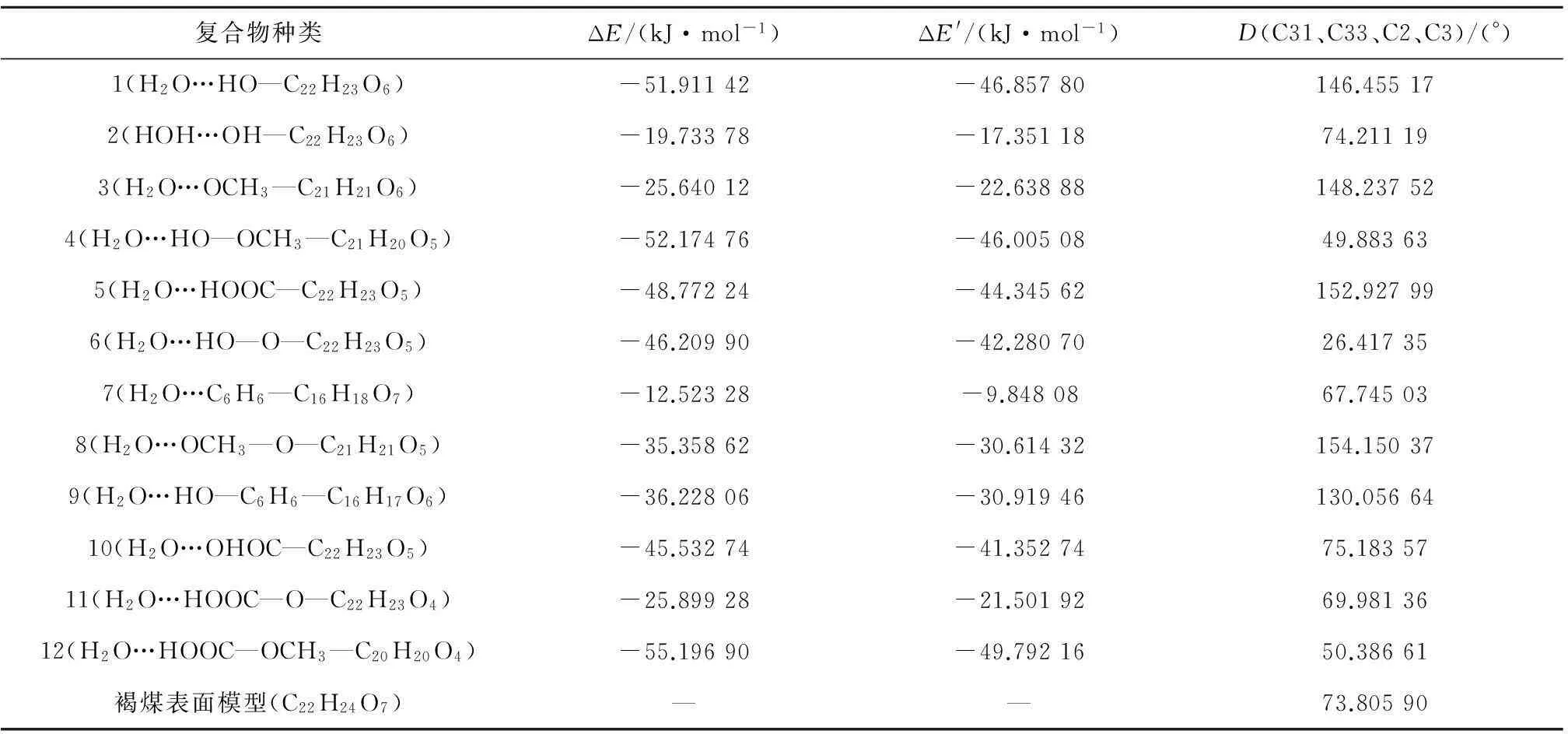
复合物种类ΔE/(kJ·mol-1)ΔE'/(kJ·mol-1)D(C31、C33、C2、C3)/(°)1(H2O…HO—C22H23O6)-51.91142-46.85780146.455172(HOH…OH—C22H23O6)-19.73378-17.3511874.211193(H2O…OCH3—C21H21O6)-25.64012-22.63888148.237524(H2O…HO—OCH3—C21H20O5)-52.17476-46.0050849.883635(H2O…HOOC—C22H23O5)-48.77224-44.34562152.927996(H2O…HO—O—C22H23O5)-46.20990-42.2807026.417357(H2O…C6H6—C16H18O7)-12.52328-9.8480867.745038(H2O…OCH3—O—C21H21O5)-35.35862-30.61432154.150379(H2O…HO—C6H6—C16H17O6)-36.22806-30.91946130.0566410(H2O…OHOC—C22H23O5)-45.53274-41.3527475.1835711(H2O…HOOC—O—C22H23O4)-25.89928-21.5019269.9813612(H2O…HOOC—OCH3—C20H20O4)-55.19690-49.7921650.38661褐煤表面模型(C22H24O7)——73.80590
从表1还可以看出,多种复合物耳朵二面角D与褐煤表面模型中的二面角D的变化范围为-50°~80°,最大偏差可达+80°,近乎直角扭曲,变形程度极大.褐煤结构变化程度不仅与形成复合物的相互作用能存在联系,也与水分子吸附的区域密切相关.如H2O吸附在复合物7上(即苯环附近区域)时,水分子与褐煤主体部分相互作用较弱,二面角D变化不大;而其与褐煤边缘区域中的羟基、羧基形成复合物时,相互作用能较大,二面角D很大,即褐煤结构扭曲变形程度越大.H2O极易被褐煤中的羟基和羧基等基团吸附,而这些含氧基团多分布在褐煤支链上,褐煤吸水后结构产生较大变化,这一结论也符合在褐煤干燥过程中结构会产生不可逆破坏的实验现象.
表2给出了12种复合物中水分子吸附区域的几何构型键长.由表2可知,复合物12中H2O吸附在2个含氧基团(—OCH3、—COOH)上,其中O54—H25和H55—O49的键长分别为0.193 nm和0.202 nm,对比复合物12形成的相互作用能,表明褐煤表面的羧基、甲氧基均与H2O形成很强的氢键作用.这一情况还可以在复合物4、复合物6、复合物8、复合物9和复合物11中发现,这些复合物中H2O与对应的活性基团的键长分别为0.185 nm、0.203 nm、0.205 nm、0.191 nm、0.203 nm和0.204 nm,也形成了氢键作用.
结合表1和表2可以发现,复合物2、复合物4、复合物6和复合物9中,H2O与褐煤中羟基的键长分别为0.198 nm、0.186 nm、0.208 nm和0.204 nm,也形成了氢键作用.不同的是复合物4、复合物6和复合物9中,H2O的一端吸附于羟基,另一端则与褐煤中其他基团存在弱相互作用,其相互作用能绝对值均大于30.919 kJ/mol;而复合物2中,H2O只与褐煤表面模型中的羟基存在相互作用,其相互作用能为-17.35 kJ/mol,这说明复合物2在这4种复合物中是最不稳定的,也表明了H2O与煤中多个不同基团或原子产生的相互作用将有助于形成稳定的煤水吸附构型.实际上褐煤侧链多、结构复杂,可以为H2O提供更多的着附点,比其他煤阶的煤更容易吸附水分子.
复合物8中,H2O吸附于羧基和甲氧基上,分别形成氢键H56…O49—C8和H55…O23—C22,键长分别为0.205 nm和0.218 nm;而复合物11中H2O则吸附在羧基和羰基上,形成氢键H56…O29—C28和H55…O24—C22,键长分别为0.204 nm和0.230 nm,这些形成的氢键都是H2O中的H与C—O或者C=O中的氧原子相互作用形成的,2种复合物的相互作用能却不尽相同,复合物8的相互作用能为-30.614 kJ/mol,而复合物11的相互作用能为-21.50 kJ/mol,这说明碳氧单键中的氧原子更能吸附到H2O,对H2O的吸附性更强.
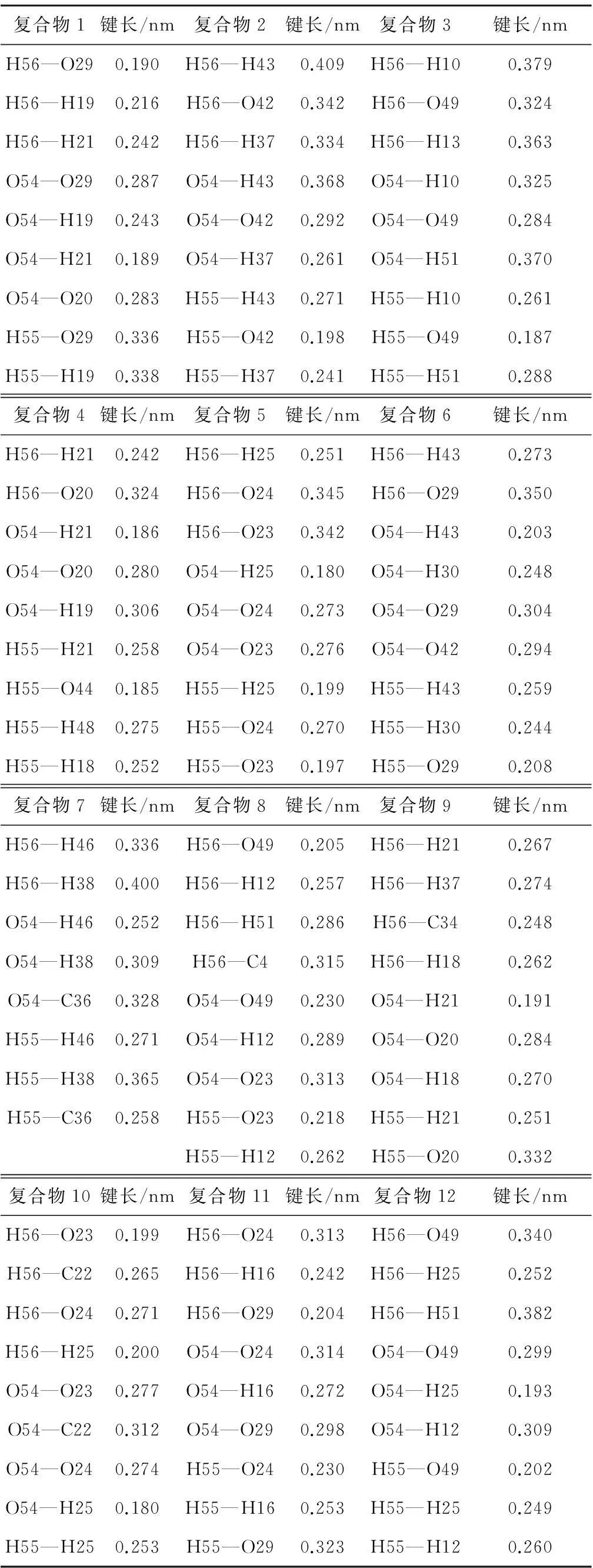
表2 水分子吸附区域的几何构型键长
12种复合物中,H2O吸附在褐煤表面上的相互作用形式除了氢键作用,其余则是范德华弱相互作用,均属于分子间弱相互作用.对比分析相互作用能和键长大小,可以发现褐煤表面区域的静电势越大(如羟基、羧基所在区域),无论正负,对H2O的吸附能越大,吸附形成键长越短,这也说明褐煤表面的静电势分布对所吸附的第一层水分子起关键作用.
2.3褐煤分子与水体系间的弱相互作用图形化分析
Johnson等[17]曾提出一种弱相互作用的可视化分析手段即RDG分析法,该方法不仅可用于氢键、静电作用和范德华作用等弱相互作用的研究,还能展现出立体位阻作用,不少学者将该方法应用到一些氢键、卤键以及配位相互作用的研究[18],其应用也得到了认同与验证,它非常直观地展示了这些弱相互作用的图像.该方法分别以空间上各点的RDG函数和sign[λ2(r)]ρ(r)函数的计算值为坐标,画出可视化的RDG填色等值图,以便直观地了解到分子中哪些区域与弱相互作用有关.RDG函数表达式为
(1)
式中:ρ(r)为随r变化的电子密度.
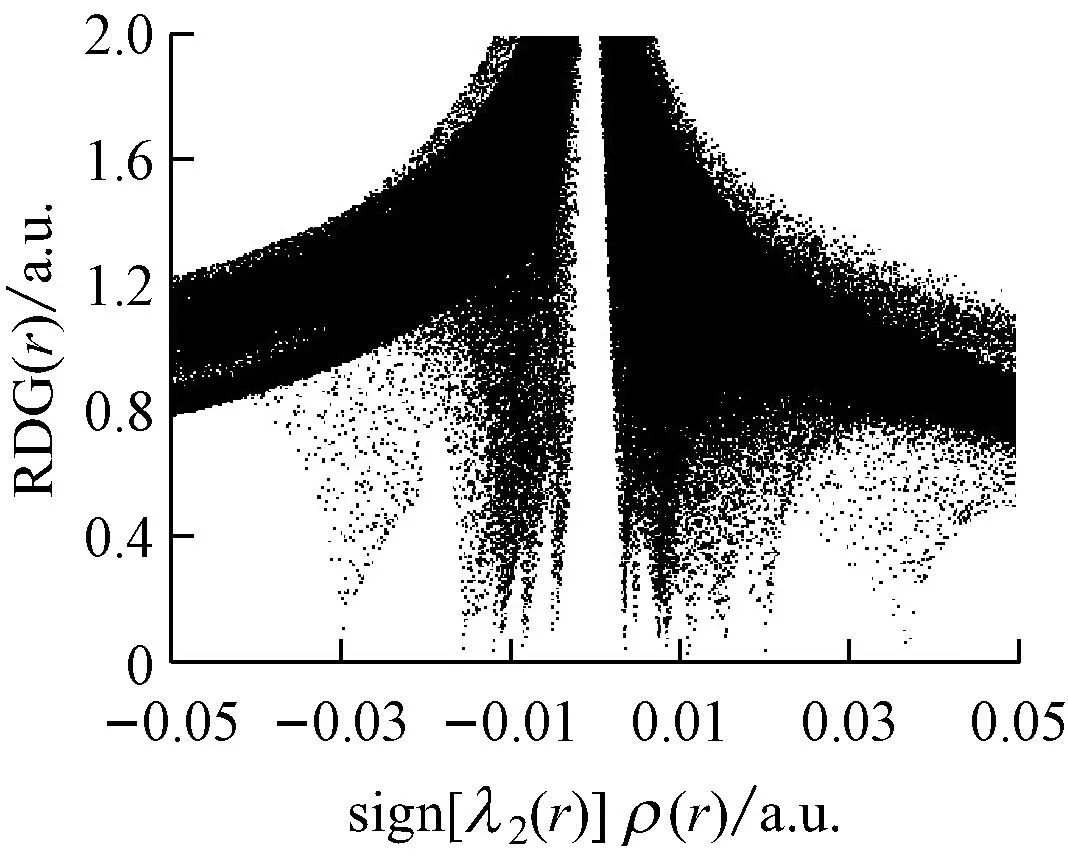
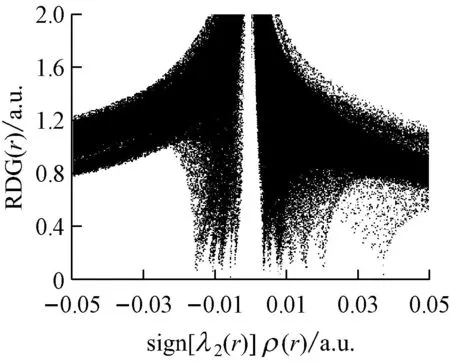
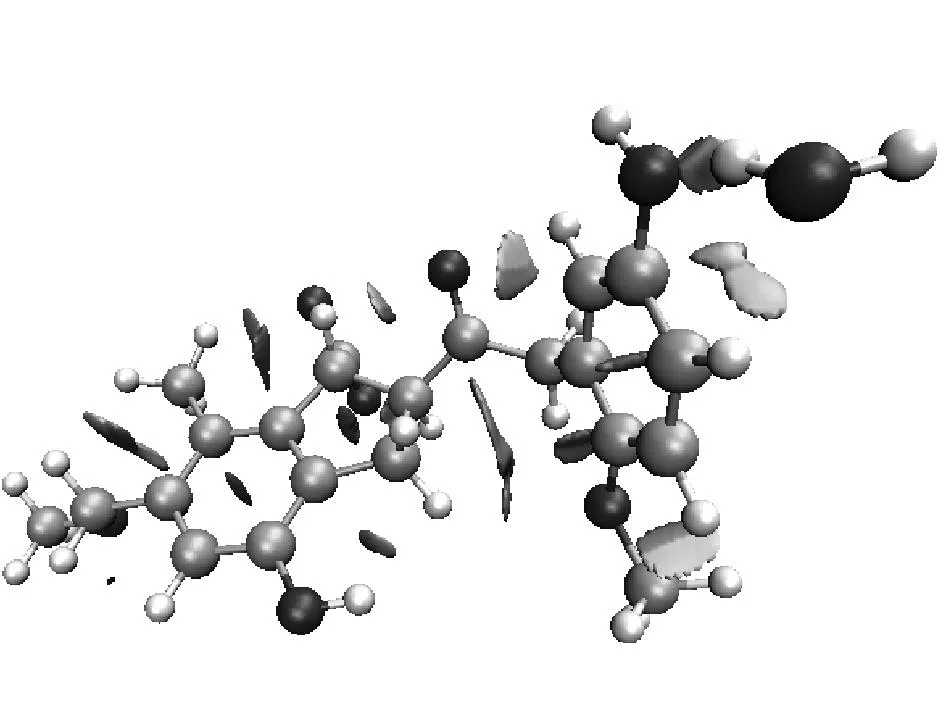
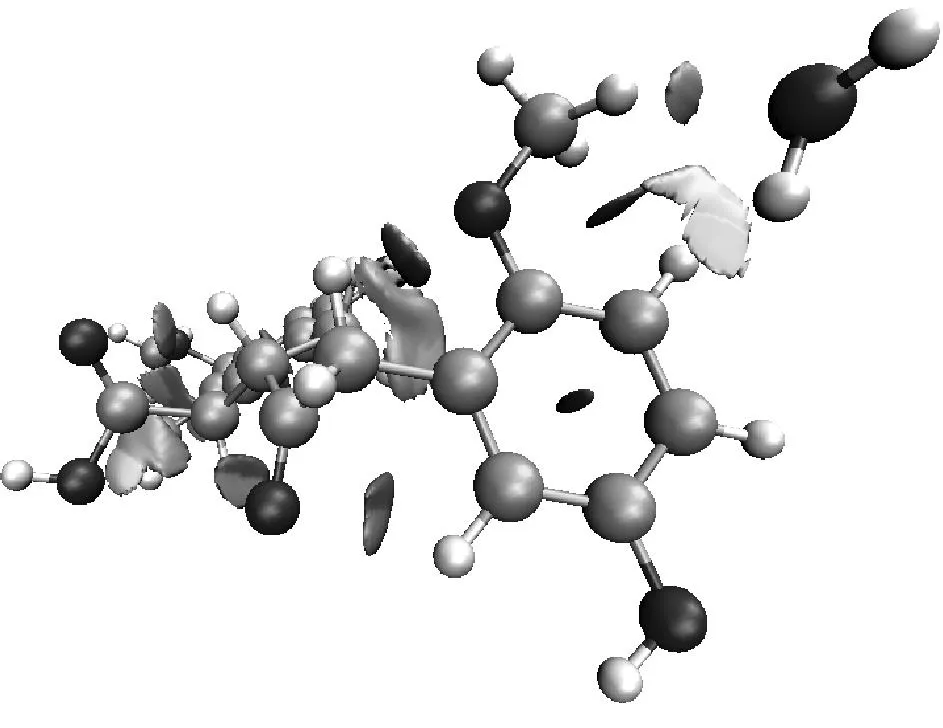
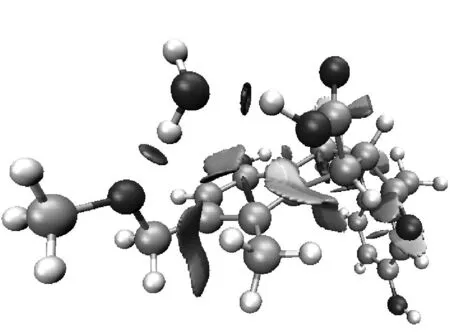
选取具有代表性的复合物2、复合物7和复合物12的RDG函数进行分析,结果见图4.其中图4(a)对应的是3种复合物的RDG-sign[λ2(r)]ρ(r)散点图,图4(b)对应的则是RDG填色等值图,图中表面颜色按照变化趋势来表现相互作用的强度,颜色越深代表相互作用越强.
复合物2
复合物7

复合物12
复合物2
复合物7
复合物12
在图4(a)中,若体系中存在弱相互作用,则此图中的左侧存在一条垂直于横轴的“突刺”,其对应的横轴数值越小,相互作用越大.由图4(a)可知,复合物2、复合物7和复合物12均存在不止一条“突刺”,即体系中H2O与褐煤表面分子形成了不止一处的相互作用,3种复合物最左侧的“突刺”垂直对应的sign[λ2(r)]ρ(r)数值分别约为-0.030 a.u.、-0.015 a.u.和-0.040 a.u.,即复合物12中的相互作用强度比其他2种复合物都要大.
图4(b)清晰地显示了3种复合物所形成的体系中存在的相互作用区域.复合物12中的填色区域颜色最深,表明此处的相互作用最强,符合图4(a)中散点图的分析结果以及之前的相互作用能和键长分析.从图4(b)还可以看出,复合物2中,H2O中的氢原子与羟基的氧原子形成氢键作用,其另一端的氧原子与苯环上的氢原子存在范德华弱相互作用;复合物7中,H2O与苯环上的碳原子和甲氧基上的氢原子都存在范德华弱相互作用;复合物12中,H2O则与羧基和甲氧基形成了氢键作用.这些分析揭示了普通键长分析忽略的范德华弱相互作用,且从RDG填色等值图显示的煤分子与水的弱相互作用体系中得到的结论,均符合目前人们对煤结构结合力以及煤与水相互作用的分子模拟的理解与研究成果.
3结论
(1) 含氧官能团的氢原子和氧原子周围区域均表现出极大的正静电势(>+104.5 kJ/mol)和负静电势(<-104.5 kJ/mol),对以静电力为主要吸附力的第一层水分子具有很强的吸附性,H2O往往先吸附到这些含氧官能团以氢键作用形成稳定的复合物.苯环和甲基等所在区域(静电势绝对值较小,为-62.7~+62.7 kJ/mol)与H2O之间存在一定的范德华弱相互作用.
(2) 褐煤表面吸附H2O的相互作用以氢键为主,其余为范德华弱相互作用,均属于分子间弱相互作用.H2O吸附在羟基和羧基上的相互作用能远大于其吸附在其他活性基团上的相互作用能,H2O吸附在苯环上形成的复合物的相互作用能绝对值最小,为-9.85 kJ/mol.褐煤表面区域羟基、羧基对H2O吸附性最强,其余的含氧基团(如醚键和甲氧基等)次之,苯环最弱.
(3) 褐煤结构复杂,在吸附H2O时,体系形成的相互作用区域并不简单地集中于某个基团或某个原子,而是与褐煤局部区域都形成一定的相互作用,从而增强了H2O的吸附作用.褐煤吸水后结构产生较大变化,其结构变化程度不仅与形成复合物的相互作用能有关,也与水分子吸附区域密切相关.
参考文献:
[1]高正阳, 李晋达, 范元周. 褐煤预干燥对锅炉传热特性及运行经济性的影响[J]. 动力工程学报, 2014, 34(3):182-188.
GAO Zhengyang, LI Jinda, FAN Yuanzhou. Influence of lignite dryness on heat transfer and operation economy of power boilers[J]. Journal of Chinese Society of Power Engineering, 2014, 34(3):182-188.
[2]LI Feng, LIU Xiangchun, SONG Lingling,etal. The effect of alkali treatment on some physico-chemical properties of Xilinhaote lignite[J]. Powder Technology, 2013, 247: 19-23.
[3]王永刚, 周剑林, 陈艳巨, 等.13C固体核磁共振分析煤中含氧官能团的研究[J]. 燃料化学学报, 2013, 41(12): 1422-1426.
WANG Yonggang, ZHOU Jianlin, CHEN Yanju,etal. Contents of O-containing functional groups in coals by13C NMR analysis[J].Journal of Fuel Chemistry and Technology, 2013, 41(12):1422-1426.
[4]虞育杰, 钟晶亮, 刘建忠. 水热提质条件下褐煤脱氧过程的量子化学研究[J]. 中国电机工程学报, 2014, 34(32): 5757-5762.
YU Yujie, ZHONG Jingliang, LIU Jianzhong. A quantum chemistry study of deoxidization process of brown coal during hydrothermal dewatering treatment[J]. Proceedings of the CSEE, 2014, 34(32): 5757-5762.
[5]虞育杰, 刘建忠, 王传成, 等. 低阶煤脱水提质技术发展现状[J]. 热力发电, 2011, 40(9): 1-4.
YU Yujie, LIU Jianzhong, WANG Chuancheng,etal.Status quo of development in dewatering for upgrading low rank coal[J].Thermal Power Generation, 2011, 40(9):1-4.
[7]王宝俊, 张玉贵, 谢克昌. 量子化学计算在煤的结构与反应性研究中的应用[J]. 化工学报, 2003, 54(4): 477-488.
WANG Baojun, ZHANG Yugui, XIE Kechang. Application of quantum chemistry calculation to investigation on coal structure and reactivity[J]. Journal of Chemical Industry and Engineering, 2003, 54(4): 477-488.
[8]王宝俊, 凌丽霞, 章日光, 等. 煤热化学性质的量子化学研究[J]. 煤炭学报, 2009, 34(9): 1239-1243.
WANG Baojun, LING Lixia, ZHANG Riguang,etal. Quantum chemistry calculation on thermochemical properties of coal[J]. Journal of China Coal Society, 2009, 34(9):1239-1243.
[9]刘晓强, 田之悦, 储伟, 等. CH4, CO2和 H2O 在非金属原子修饰石墨烯表面的吸附[J]. 物理化学学报, 2014, 30(2):251-256.
LIU Xiaoqiang, TIAN Zhiyue, CHU Wei,etal. CH4, CO2and H2O adsorption on nonmetallic atom-decorated graphene surfaces[J]. Acta Physico-Chimica Sinica, 2014, 30(2):251-256.
[10]曾凡桂, 贾建波. 霍林河褐煤热解甲烷生成反应类型及动力学的热重-质谱实验与量子化学计算[J]. 物理化学学报, 2009, 25(6):1117-1124.
ZENG Fangui, JIA Jianbo. Reaction types and kinetics of methane generation from Huolinhe lignite pyrolysis by TG/MS experiment and quantum chemical calculations[J]. Acta Physico-Chimica Sinica, 2009, 25(6):1117-1124.
[11]MATHEWS J P, CHAFFEE A L. The molecular representations of coal-A review[J]. Fuel, 2012, 96(1): 1-14.
[12]WENDER I. Catalytic synthesis of chemicals from coal[J]. Catalysis Reviews-Science and Engineering, 1976, 14(1): 97-129.
[13]BENZIE I F F, SZETO Y T. Total antioxidant capacity of teas by the ferric reducing/antioxidant power assay[J]. Journal of Agricultural and Food Chemistry, 1999, 47(2): 633-636.
[14]LU T, CHEN F. Multiwfn: a multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 2012, 33(5): 580-592.
[15]HUMPHREY W, DALKE A, SCHULTEN K. VMD: visual molecular dynamics[J]. Journal of Molecular Graphics, 1996, 14(1): 33-38.
[16]RAO Zhonghao, ZHAO Yuemin, HUANG Congliang,etal. Recent developments in drying and dewatering for low rank coals[J]. Progress in Energy and Combustion Science, 2015, 46: 1-11.
[17]JOHNSON E R, KEINAN S, MORI-SANCHEZ P,etal. Revealing noncovalent interactions[J]. Journal of the American Chemical Society, 2010, 132(18): 6498-6506.
[18]许惠英, 王维, 邹建卫. PH2X与五元杂环体系磷键相互作用的理论研究[J]. 化学学报, 2013, 71(8): 1175-1182.
XU Huiying, WANG Wei, ZOU Jianwei. Theoretical study of pnicogen bonding interactions between PH2X and five-membered heterocycles[J]. ACTA CHIMICA SINICA, 2013, 71(8):1175-1182.
Micro-mechanism of Water Molecule Adsorption on Lignite Surfaces
GAOZhengyang1,LÜShaokun1,LIJinda2,YANGPengfei1,CHENChuanmin3
(1. School of Energy, Power and Mechanical Engineering, North China Electric Power University,Baoding 071003, Hebei Province, China; 2. Guangdong Yudean Jinghai Power Generation Co., Ltd., Jieyang 515223, Guangdong Province, China; 3. College of Environmental Science and Engineering, North China Electric Power University, Baoding 071003, Hebei Province, China)
Abstract:The micro-mechanism of water molecule adsorption on lignite surfaces was investigated by using density functional theory, following which different lignite surface models and coal-water adsorption models were obtained through calculation by Gaussian09 program at the level of B3LYP/6-311G+(d,p), while the interaction energies were corrected by complete counterpoise method considering the basis set superposition error (BSSE). Moreover, graphical analyses were also conducted for different lignite surface models using electrostatic potential diagram, scatter diagram and reduced density gradient (RDG) color-filled isosurface map with Multiwfn program and VMD software. Results show that the water adsorption on lignite surface is of the weak interaction physical adsorption kind, the major part of which is hydrogen bonding interaction while the rest is Van Edward interaction. The adsorbability of hydroxyl and carboxyl to water is the strongest, followed by other oxygen functional groups such as the ether bond and methoxy, etc., and that of benzene ring is the weakest. The interaction region formed through water adsorption does not simply concentrate on some groups or an atom but somewhat interacts with local lignite areas. Thus the water adsorption is reinforced and the structure of lignite is impacted obviously.
Key words:lignite surface; water molecule; hydrogen bond; density functional theory; RDG color-filled isosurface map
文章编号:1674-7607(2016)04-0258-07中图分类号:O641.12+1
文献标志码:A学科分类号:150.30
猜你喜欢
高中数理化(2022年14期)2022-08-15
科技创新与生产力(2021年11期)2021-12-30
波谱学杂志(2021年3期)2021-09-07
科教新报(2021年11期)2021-05-12
原子与分子物理学报(2019年5期)2019-04-28
信阳师范学院学报(自然科学版)(2018年1期)2018-08-09
意林原创版(2017年11期)2017-12-01
中学化学(2015年12期)2016-01-19
原子与分子物理学报(2014年3期)2014-02-28
