
用振动力学有限元分析方法估算多类烃的液体热导率
2016-08-06 07:11仇明华刘万强陈冠凡刘凤萍
化工学报 2016年7期
仇明华,刘万强,陈冠凡,刘凤萍,岳 明
研究论文
用振动力学有限元分析方法估算多类烃的液体热导率
仇明华1,2,刘万强1,2,陈冠凡1,刘凤萍1,岳明1
(1湖南科技大学化学化工学院;2理论有机化学与功能分子教育部重点实验室,湖南 湘潭 411201)
摘要:将烃分子结构模拟成弹性体,用振动力学有限元分析方法对分子 3D结构进行有限元分析,建立分子结构体系的刚度矩阵和多自由度振动方程,用 MATLAB软件包求解得到分子结构固有频率和刚度矩阵特征值,将分子结构固有频率和刚度矩阵特征值作为参数,用多元回归方法建立液态烃类热导率QSPR模型。该模型对训练集581个多类烃的液态热导率的计算值与实验值的相关系数为0.9874,平均绝对误差小于0.00259 W·m-1·K-1,平均相对误差小于2.39%;对测试集22个烃类化合物热导率的预测值与实验值的相关系数为0.9550,平均绝对误差小于0.00263 W·m-1·K-1,平均相对误差小于2.42%。该计算模型的计算值和实验吻合,可用于链烃、烯烃、炔烃及单环烷烃、多环烷烃、萘烷、芴烷、菲烷、茚烷、蒎烷等复杂结构烷烃的液体热导率的计算。
关键词:热导率;热传导;热力学性质;分子结构参数;有限元分析
引 言
热导率是化工、生物、冶金、机械、石油和能源等工程领域传热设计中需要的重要基础数据,也是采暖、保温、传热、散热、制冷等工程中寻找合适热交换器的重要依据,同时也是化合物最重要的物理性质之一。由于有机物种类繁多,如果通过实验测定工作量太大,有些化合物实测起来也许非常困难。因此,寻找化合物热导率数据更加准确和简便的计算方法一直是化学和化工领域研究的重点课题之一。
从已有的热导率的计算方法[1-19]来看,可分为基于分子运动理论方法和基团贡献法等。这些方法大多都离不开对化合物其他物理性质的依赖,如Latini法、Sastri方法和Teja方法[1-3],都是在对比温度基础上进行估算的。有些方法是以热导率实验值为前提的,如后续的基团贡献法、拓扑指数法、神经网络法和伪格法等[4-8]。同时为了减小热导率计算值与实验值的误差,对计算方法的约束条件也越来越多,如Arikol和Gurbuz方法[10],Dana和Dabir[11]方法等需要温度、临界温度、临界压力、分子量和标准沸点等。
因此探索从分子结构直接计算热导率(不依赖化合物的其他实验数据和物理性质)是非常有意义的工作。这方面的研究始于Jamieson等[12-13],在给出一个估算λL的通用方程后,再分析分子结构和分子大小对方程中常数的影响。张克武等[14]基于气体不平衡理论方程推导出液体热导率估算方程,同样也是将分子结构的影响归结为不同碳原子数、不同化学键和不同基团对方程系数的影响,并将温度的影响通过临界温度和熔点划分为不同的温度段,使纯质液体热导率的估算误差远远小于Latini方法的误差。直接用分子结构参数来计算纯质液体热导率的研究始于文献[15-17]。仇明华等[15]以4个基团参数、1个极化效应指数PEI和1个温度参数,对45个链烷烃分子的155个热导率数据建立了高相关性估算模型。曹晨忠等[16]采用了拓扑量子方法,以键邻接矩阵特征根为参数,对45种链烷烃在不同温度下的155个热导率数据进行回归,建立了4参数拟合方程。聂长明等[17]从链烷烃分子中各原子的电负性效应、支化效应及空间效应出发,构建了结构拓扑指数PXm,对液相链烷烃在不同温度的155个热导率数据点建立了4参数的定量构效关系模型。此外万丽华等[18-19]采用EMD方法Green-Kubo理论采用动力学模拟客体分子数对烃类水合物热导率的影响。尽管上述方法有所突破,但理论不够具体,经验性非常明显,因而局限了估算方程的应用范围。
物质热量传递机理[20]认为液体分子处在其周围相邻分子形成的“分子笼”中,通过分子的运动(分子、原子或电子的平动、振动和转动)来传递动量和能量。然而由于人们对液体分子运动仍然不能精确描述,同时分子可能以某种形式储存能量等原因,对液体分子的运动和热传导仍然不能用公式准确描述。空间刚架元分析[21-22]是机构振动力学分析的有效手段之一。它将一个结构看成是由多个空间杆元和节点构成的体系。其中的空间杆元是没有质量的弹性体,体系的整体质量分散在各节点上,这与分子结构非常契合。在分子中,化学键就相当于一个没有质量的空间杆元,也可以认为是一个没有质量的弹性体,原子或基团就相当于杆元的节点,是整个分子质量的分散点。这与物质通过内部原子、分子的振动和相互碰撞进行热量传导的导热机理吻合。
利用弹性细杆非线性力学方法中的DNA力学模型理论,可以计算每个化学键空间杆元的6个参数,即弹性模量、剪切弹性模量、横截面积、长度、惯性矩和极惯性矩。求出整体刚度矩阵的特征值和多自由度分子结构体系振动方程的特征根——固有频率。可以用各阶特征值的平均绝对值和各阶固有频率之和作为定量构效关系(QSPR)模型参数,应用于液态烃类热导率的计算。
1 理论与方法
1.1 分子结构建立和优化
用空间刚架元分析分子结构时,首先确定刚架元节点的空间3D坐标,即烃类分子中CHn基团的3D坐标的确定。本文首先用Chem3D绘制分子的3D结构,再导入到Gaussian 09软件,用RHF方法,6-31G基组,优化分子结构,同时使用freq关键词计算频率,看有无虚频以判断优化后构型是否为最优构型。最后得到分子中原子的空间坐标。
1.2 化学键空间刚架元系数的定义与计算
本文将隐氢分子图中的化学键定义为化学键空间刚架元。这样烃类分子隐氢图中只涉及 C—C单键、CC双键和CC三键,其键长L可以根据输入的分子的3D坐标计算由MATLAB有限元分析程序得到[19-20]。分子空间刚架元的其余5个系数:弹性模量(E)、剪切弹性模量(G)、横截面积(A)、惯性矩(Ix和 Iy)、极惯性矩 Iz的值分别见表 1,其中Ix=Iy。有关6个系数的定义和计算可参见文献[21-23]。
1.3 分子结构刚度矩阵平均绝对特征值δm的定义与计算
在定义和计算得到化学键空间刚架元6个系数之后,本文建立分子结构的整体刚度矩阵[21-23]。先建立单个化学键的单元刚度矩阵ki,最后建立分子整体刚度矩阵K。K是一个6n×6n阶方阵,这里的n是指分子隐氢图中的原子基团数,即节点数。如果隐氢分子图中的原子基团(节点)数目越多,其整体刚度矩阵的阶数就越高,在求解特征值时,可以使用MATLAB软件求解,其程序代码为“eig”,即
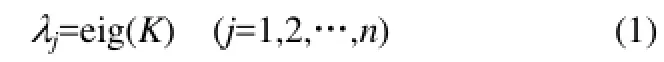
本文定义一个分子结构刚度矩阵平均绝对特征值,用“δm”表示,即

式中,|λj|为各阶特征值的绝对值,以保证各阶特征值在求和过程中不被正负抵消;nc为分子中的节点数,即隐氢分子图中的原子或基团数,对于烃类分子而言,就是分子中的碳原子数。
表1 C—C单键、CC双键和CC三键空间刚架元的6个参数值Table 1 Values of parameters for C—C, CC and CC bond

表1 C—C单键、CC双键和CC三键空间刚架元的6个参数值Table 1 Values of parameters for C—C, CC and CC bond
Elastic Shear elastic Type of Moment of Cross-sectional Moment of Polar moment of modulus (E) Bond modulus (G) area (A) inertia (Ix) inertia (Iy) inertia (Iz) C—C 0.8322 0.2774 3.1412 0.7853 0.7853 1.5706 C C 1.4688 0.4896 2.3078 0.4239 0.4239 0.8478 C C 2.0108 0.6703 1.9023 0.2895 0.2895 0.5790
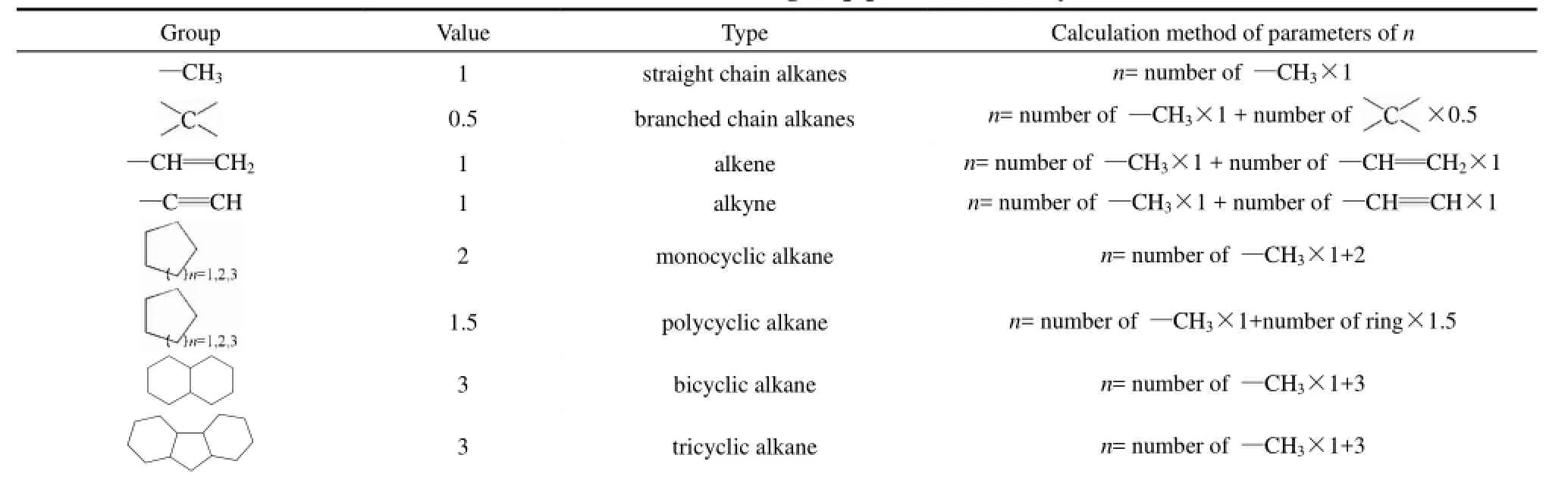
表3 烃类分子中特征基团参数n的计算方法Table 2 Calculation methods of n values for group parameters of hydrocarbon molecules
1.4 分子结构固有频率计算
从文献[22]可知,多自由度振动体系的特征方程为

由此方程计算 p2的n个正实根p2i,并按 pi≤ pi+1(i=1,2,…,n)排列。pi称为系统的固有频率,其中不等于零的最小固有频率称为基频。式(3)中的K是分子结构体系的整体刚度矩阵,M是分子结构体系的集中质量矩阵,即由隐氢分子图中各原子基团的质量构成。固有频率的计算可直接调用MATLAB程序代码“eig”求解,即

根据以上方法计算得到 78个烃分子固有频率pi和各阶固有频率之和(∑pi)的值。
1.5 分子结构特征基团参数n的定义与计算
对78个烃分子的581个热导率实验数据的回归分析发现,同系列烃分子中某些特征基团可具有相同的取值,如分子中的甲基可取值为 1,季碳原子可取值为0.5,端基烯、炔键可取值为1,单环烷烃中的环结构可取值为 2,多环烷烃的环结构可取值为1.5,分子中的十氢化萘、芴结构可取值为3。特征基团参数n的定义、取值及计算方法列于表2。
特征基团参数n的计算方法如下所示。
(1)烷烃:在烷烃分子中,只有甲基—CH3和碳链中季碳原子作为分子中的特征基团,其余碳链中间的—CH2—,碳原子不是特征基团,其结构特征在键长数据L中已经表征。特征基团参数n的计算方法如下。
例1:2,2,5,5-四甲基己烷,分子含6个甲基,2个季碳原子,其特征参数n值的计算结果为

(2)烯烃:只有处于端基的烯键—CH═CH2符合特征基团,不处于链端的双键结构不属于特征结构,其结构特征在键长数据L中已经表征。
例2:2-甲基-1,3-丁二烯分子,虽然有两个烯键,但只有1个烯键符合,所以对应的特征参数n值仅由1个甲基数和1个烯键数来确定,即
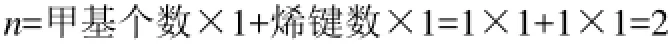
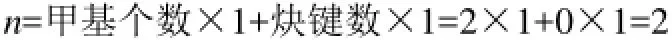
(4)环烷烃:分子中的单环结构均作为特征基团计算。
例 4:乙基环己烷分子,分子中的特征基团为1个甲基,1个C6环,特征基团参数n为

(5)环烃:
例5:以双-(2,4,6-环己基)甲烷分子为例,分子中的特征基团有6个甲基,2个环己基,所以该分子的特征基团参数n值为9,即

(6)复杂烷烃:特征基团包括表2列出的8类结构的一种或几种。
例6:α-甲基十氢化萘分子中有1个甲基和1个十氢化萘环结构,特征基团参数n计算如下
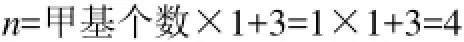
1.6 QSPR模型的建立
最佳子集回归算法(best subsets regression)是寻找特定数目自变量组合中最佳的一种或几种搭配的多元线性回归方法。可以根据使用者设定的条件选择含有1,2,3,…,n个变量的最佳变量组合。该方法可以逐步考察不同变量组合对因变量的影响。因此本文采用最佳子集回归算法筛选参数并建立多元线性回归(multiple linear regression,MLR)方程。
1.6.1 热导率与温度的关系 由于分子的热导率与温度的关系非常密切,在建立QSPR模型时既要考虑分子结构的影响,也要考虑到温度的影响。文献[12]对于热导率与温度的关系作了专门研究,得到的关联方程为
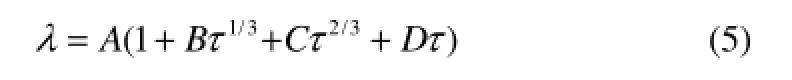
式中,τ=1-T/Tc,这里的Tc是临界温度,A是临界热导率,B、C、D是常数。由此可以看出,温度对液体热导率的关系是一种非线性关系。由于式(5)的表达依赖于临界温度,本文并不直接将该式引入QSPR模型,而是曲线拟合,发现烃类液体热导率受温度影响的函数表达式可归结为

式中,T是热力学温度,a为回归常数,b、c为参数的回归系数。1.6.2 结构参数筛选及QSPR方程建立 将前文得到结构参数∑pi,δm,n,以及其他结构参数组成参数集,将化合物300 K热导率作为因变量,通过最佳子集回归方法筛选分子结构参数,得到固有频率之和∑pi(简称总频)、整体刚度矩阵平均绝对特征值δm及组成的最佳3参数回归方程式(7)
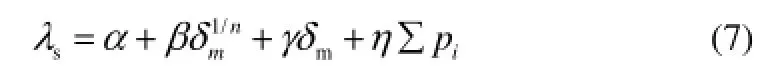
再考虑温度对热导率的影响,将式(6)和式(7)合并,得到式(8)
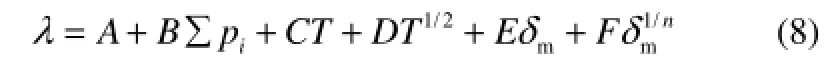
式(8)为本文建立的多类烃液体热导率的QSPR模型。
2 结果与讨论
2.1 模型的拟合
本文从文献[24]收集了多类烃(直链烷烃分子20个,支链烷烃分子20个,烯烃分子4个,炔烃分子3个,环烷烃分子18个,萘烷分子12个,芴分子1个)的581个液体热导率实验数据,作为训练集,拟合QSPR方程式(8),得到最终的热导率QSPR方程式(9)
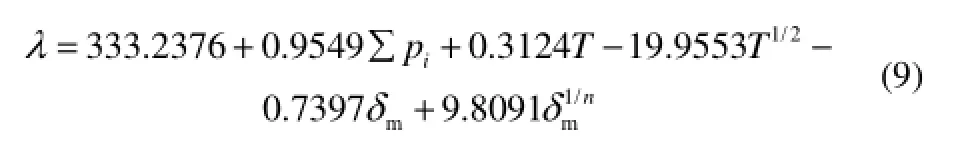
(train set: n=581, R=0.9874, s=3.45, F=4483;
test set: n=22, R=0.9550, s=4.42, F=207)
根据式(9)计算得到 581个化合物的热导率数据,该方程对581个训练集热导率的计算值与实验值的相关系数为0.9874,平均相对误差为2.39%。实验值与计算值的相关性如图1所示。从图1可以看出,实验值和计算值数据点分布基本在对角线上,表明实验值和计算值基本一致。

图1 热导率计算值与实验值比较Fig.1 Plot of experimental λexp.versus calculated λcalc.
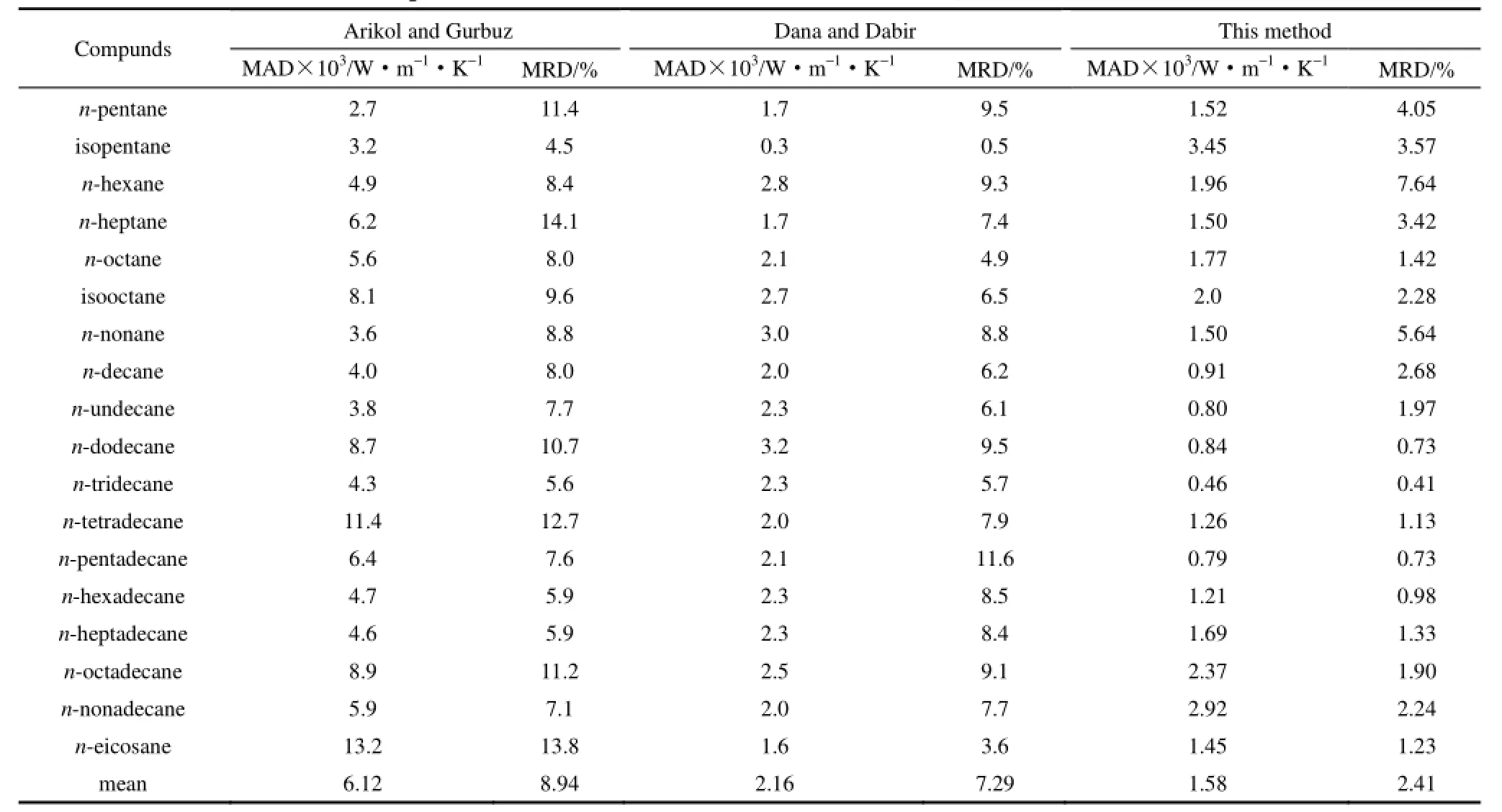
表3 Arikol and Gurbuz, Dana and Dabir和本文方法预测结果的比较Table 3 Comparison of new method with Arikol and Gurbuz, Dana and Dabir methods
2.2 模型方程的检验和方程预测性能力分析
为了进一步验证模型(9)预测能力,从文献[24]收集22个烃的液体热导率数据作为测试集,用式(9)预测了这些物质的热导率,预测值和实验值的相关系数R=0.9550,平均相对误差为2.42%。预测结果在实验值的误差范围之内(5%)。预测值和实验值的比较如图1所示。说明式(9)具有良好的预测能力。
2.3 估算结果比较
为了考察本文方法与已有方法的优劣,随机选取了18个烃,用不同的方法计算这些有机物的热导率。一种方法为Arikol等[10]提出的热导率关联式,该关联式在已知温度、临界温度、临界压力、分子量和标准沸点的条件下,计算了液体热导率。另一方法是Dana等[11]提出的热导率关联式。表3同时列出了Arikol、Dana和本文方法的计算结果。
从表3可以看出,本文提出的计算方程的平均绝对误差为0.00158 W·m-1·K-1,平均相对误差为 2.41%;Arikol 和Gurbuz法[10]的平均绝对误差为0.00612 W·m-1·K-1,平均相对误差为8.94%;Dana和 Dabir法[11]的平均绝对误差为 0.00216 W·m-1·K-1,平均相对误差为 7.29%。本文方法计算结果明显优于其他两种方法。特别是平均相对误差优势更为明显。同时表 3的数据中,正辛烷(n-octane)和异辛烷(isopentane)两个烷烃不在本文的训练集样本中,是拟合集以外的预测结果。式(9)对正辛烷的预测结果明显优于其他两种方法的估算结果。对异戊烷所产生的绝对误差与Arikol和Gurbuz法接近,相对误差仍然小于 Arikol和Gurbuz法,但比Dana和Dabir法的误差略大。
2.4 热导率的影响因素
2.4.1 特征基团对液态烃热导率的影响 在式(9)中,对指数项的回归系数为9.8091,由于平均绝对特征值(δm)总是大于1的数值。因此,所有增加n值的因素都使得指数项的值变小,说明用振动力学有限元方法提取的结构参数δm高估了液态烃分子的热导率,必须通过指数项(1/n)来加以修正。
2.4.2 甲基的影响 在以化学键空间刚架元为结构单元提取分子结构特征参数时,落在节点处的基团可分为两种类型,一类是化学键的端点,另一类是两个或两个以上化学键的连接点。对于烃分子而言,甲基属于端点基团,由于有一端与另一化学键连接,除了振动以外,还可以发生转动,这种转动不利于分子间热量传导,同时相同碳原子数的分子,甲基越多,支链越多,分子主链越短,影响了分子能量传递效率。因此,分子中的甲基数越多,与碳原子数相同的直链烃比较,其热导率越小。如正癸烷在320 K时的液体热导率值为0.125 W·m-1·K-1,而 2,2,5,5-四甲基己烷的液体热导率值为 0.092 W·m-1·K-1。
3 结 论
本文用振动力学有限元分析方法,提取的分子结构特征参数和基团特征参数能定量地表达分子结构对多类液态烃热导率的影响规律。得到的定量构效关系方程为:λ=333.2376+0.9549∑pi+0.3124T-19.9553T1/2-0.7397δm+9.8091。
该方程适用于从链烷烃、烯烃、炔烃到单环烷烃、多环烷烃、十氢化萘类、芴烷类、茚烷类、菲烷类、蒎烷类等复杂结构烃类的液态热导率的估算。方程中的结构参数固有频率 pi和平均绝对特征值δm,无须依赖其他实验数据或临界参数。用固有频率和刚度矩阵特征值作为结构参数来定量描述分子的液体热导率随分子结构不同的变化规律,与物质通过内部原子、分子的振动和相互碰撞进行热量传导的导热机理吻合。同时在方程中引入分子结构特征基团参数n的意义重大,它不仅揭示了分子结构的差异对分子宏观性能的影响,而且扩大了预测方程的适用范围,使估算方程可以推广到其他复杂有机分子液体热导率的预测,为工程应用中热导率数据提供了比较可靠的计算方法。
References
[1] DRAGHI N C, FAYET G, CRETON B, et al. A general guidebook for the theoretical prediction of physicochemical properties of chemicals for regulatory purposes[J]. Chem. Rev., 2015, 115(24): 13093-13164.
[2] SASTRI S S, RAO K K. A new temperature-thermal conductivity relationship for predicting saturated liquid thermal conductivity[J]. Chem. Eng. J., 1999, 74(3): 161-169.
[3] PATEL N C, TEJA A S. A new cubic equation of state for fluids and fluid mixtures[J]. Chem. Eng. Sci., 1982, 37(3): 463-473.
[4] CHEN Q L, WU K J, HE C H. Thermal conductivity of ionic liquids at atmospheric pressure: database, analysis, and prediction using a topological index method[J]. Ind. Eng. Chem. Res., 2014, 53(17): 7224-7232.
[5] HEZAVE A Z, RAEISSI S, BOLOOKI M L. Estimation of thermal conductivity of ionic liquids using a perceptron neural network[J]. Ind. Eng. Chem. Res., 2012, 51(29): 9886-9893.
[6] CARRETE J, TRINIDAD M M, MANUEL G, et al. Thermal conductivity of ionic liquids: a pseudolattice approach[J]. J. Phys. Chem., 2012, 116(1): 1265-1273.
[7] LAZZÚS J A. A group contribution method to predict the thermalconductivity λ(T,P) of ionic liquids[J]. Fluid Phase Equilibria, 2015, 405(11): 141-149.
[8] POLING B E, PRAUSNITZ J M, CONNELL J P. The Properties of Gases and Liquids[M]. 5th ed. Boston: The McGraw-Hill Companies, Inc., 2001: 420-436.
[9] WATANABE H, KATO H. Thermal conductivity and thermal diffusivity of twenty-nine liquids: alkenes, cyclic (alkanes, alkenes, alkadienes, aromatics), and deuterated hydrocarbons[J]. J. Chem. Eng. Data, 2004, 49(4): 809-825.
[10] ARIKOL M, GURBUZ H. A new method for predicting thermal conductivity of pure organic liquids and their mixtures[J]. Can. J. Chem. Eng., 1992, 70(6): 1157-1163.
[11] DANA M K, DABIR S V. A correlation for the prediction of thermal conductivity of liquids[J]. Ind. Eng. Chem. Res., 1998, 37(5): 2064-2068.
[12] JAMLESON D T. Thermal conductivity of liquids[J]. J. Chem. Eng. Data, 1979, 24(3): 244-246.
[13] JAMIESON D T, CARTWRIGHT G. A correlation for the prediction of thermal conductivity of liquids[J]. J. Chem. Eng. Data, 1980, 25(3): 199-201.
[14] 张克武, 张玉英. 液体烃的分子结构与导热率[J]. 化工学报, 1999, 50(2): 247-252.
ZHANG K W, ZHANG Y Y. Extended application of TENSG on thermal conductivity and molecular structure of liquid hydrocarbons [J]. Journal of Chemical Industry and Engineering (China), 1999, 50(2): 247-252.
[15] 仇明华, 曹晨忠. 液体链烷烃热导率与分子结构的关系[J]. 湘潭大学自然科学学报, 2002, 24(3): 69-73.
QIU M H, CAO C Z. Relationship between the thermal conductivity of liquid alkane and it’ s molecular structure[J]. Natural Sci. J. Xiangtan University, 2002, 24(3): 69-73.
[16] 高硕, 曹晨忠. 拓扑量子方法估算液体链烷烃热导率[J]. 物理化学学报, 2006, 22(16): 1478-1483.
GAO S, CAO C Z. A topological quantum method for the estimation of the thermal conductivity of liquid alkanes[J]. Acta. Phys. Chim. Sin., 2006, 22(16): 1478-1483.
[17] 彭国文, 肖方竹, 聂长明, 等. 液相链烷烃热导率与其结构定量关系[J]. 化工学报, 2011, 62(3): 604-610.
PENG G W, XIAO F Z, NIE C M, et al. Quantitative relationship between thermal conductivity and structure of liquid alkanes[J]. CIESC Journal, 2011, 62(3): 604-610.
[18] 万丽华, 梁德青, 吴能友, 等. 客体分子数对甲烷水合物导热性能影响的分子动力学模拟[J].化工学报, 2012, 63(2): 382-386.
WAN L H, LIANG D Q, WU N Y, et al. Molecular dynamics simulation of gest molecule number on methane hydrate thermal performance[J]. CIESC Journal, 2012, 63(2):382-386.
[19] 万丽华, 梁德青, 关进安. 烃类水合物导热特性的分子动力学模拟[J]. 化工学报, 2014, 65(3): 792-796.
WAN L H, LIANG D Q, GUAN J A. Characteristic of thermal conduction in hydrocarbon hydrates using molecular dynamics method[J]. CIESC Journal, 2014, 65(3): 792-796.
[20] 马庆芳. 传热学[M]. 北京: 人民教育出版社, 1976.
MA Q F. Heat Transfer [M]. Beijing: People’s Educational Press, 1976.
[21] 刘延柱. 弹性细杆的非线性力学—DNA力学模型的理论基础[M].北京: 清华大学出版社, 2006.
LIU Y Z. Nonlinear Mechanics of Thin Elastic Rod: Theoretical Basis of Mechanical Model of DNA[M]. Beijing: Tsinghua University Press, 2006.
[22] 方同, 薛璞. 振动理论及应用[M]. 西安:西北工业大学出版社, 2002: 99.
FANG T, XUE P. Vibration Mechanics and Application[M]. Xi’an: NWPU Press, 2002: 99.
[23] KATTAN P I. MATLAB Guide to Finite Element Analysis[M]. Berlin: Springer-Verlag, 2003.
[24] VARGAFTIK N B, FILIPPOV L P, TARZIMANOV A A, et al. Handbook of Thermal Conductivity of Liquids and Gases[M]. Boca Raton FL: CRC Press, 1994.
2015-10-13收到初稿,2016-01-23收到修改稿。
联系人:刘万强。第一作者:仇明华(1958—),男,教授。
Received date: 2015-10-13.
中图分类号:O 642.1;TQ 013.1
文献标志码:A
文章编号:0438—1157(2016)07—2672—07
DOI:10.11949/j.issn.0438-1157.20151546
基金项目:国家自然科学基金项目(21472040, 21202043);湖南省教育厅科学研究项目(13C302)。
Corresponding author:LIU Wanqiang,wanqiangliu@foxmail.com supported by the National Natural Science Foundation of China (21472040, 21202043) and the Science Research Project of Hunan Provincial Department of Education(13C302).
Conductivity estimates of liquid hydrocarbons by finite element analysis in vibration mechanics
QIU Minghua1,2, LIU Wanqiang1,2, CHEN Guanfan1, LIU Fengping1, YUE Ming1
(1School of Chemistry and Chemical Engineering, Hunan University of Science and Technology;2Key Laboratory of Theoretical Organic Chemistry and Functional Molecule, Ministry of Education, Xiangtan 411201, Hunan, China)
Abstract:Given hydrocarbon molecular structures as elastomers, three dimentional (3D) molecular structure of hydrocarbons was analyzed by finite element analysis in vibration mechanics. The stiffness matrix and vibration equation of molecular structure system was established, and its eigen values of inherent frequency and stiffness matrix were obtained by calculation using MATLAB software, followed by QSPR model for the liquid hydrocarbon conductivity by multivariate regression method. For training set consisting of 581 liquid hydrocarbons, the correlation coefficient R between the calculated values of liquid conductivity using QSPR model and the experimented data is 0.9874, the mean absolute error less than 0.00259 W·m-1·K-1, and the relative error less than 2.39%. For testing set comprising 22 liquid hydrocarbons, the correlation coefficient R is 0.9550, the mean absolute error less than 0.00263 W·m-1·K-1, and the relative error less than 2.42%. It showed that the calculated values by this QSPR model fit experimental data well in terms of conductivity of liquid hydrocarbon. It can be used to estimate the liquid conductivity of complex hydrocarbons such as acyclic alkanes, alkenes, alkynes, monocycle alkanes, polycyoalkanes, decahydronaphthalenes, fluorene alkanes, perhydrophenanthrenes, indene alkanes, and pinanes.
Key words:conductivity; heat conduction; thermodynamic properties; molecular structure descriptor; finite element analysis
猜你喜欢
储能科学与技术(2022年5期)2022-05-10
数学物理学报(2022年1期)2022-03-16
红蜻蜓·中年级(2021年12期)2021-12-19
装备制造技术(2020年2期)2020-12-14
数学物理学报(2020年5期)2020-11-26
陶瓷学报(2020年5期)2020-11-09
哈尔滨师范大学自然科学学报(2017年1期)2017-06-23
文艺生活·中旬刊(2016年11期)2016-12-13
航空兵器(2016年4期)2016-11-28
科学与财富(2016年15期)2016-11-24
