
DNA序列特征提取与功能预测技术的探讨
2016-11-16 14:24谭生龙
电脑知识与技术 2016年25期
关键词:特征提取
谭生龙
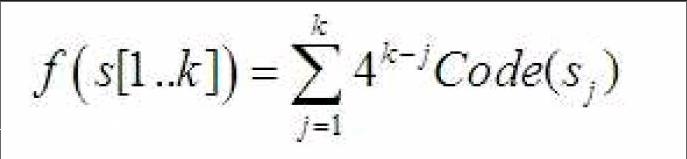
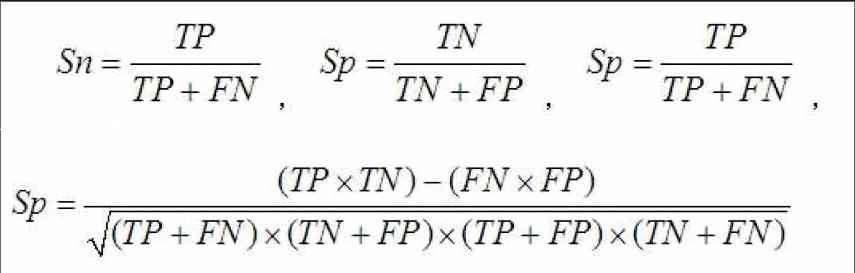
摘要:测序技术的快速进步产出了大量生物序列,DNA序列是生物大数据的重要组成部分,仅有极少部分DNA序列已通过实验验证了功能;通过机器学习方法快速预测DNA序列的功能是确实可行的途径。本文探讨了将DNA序列转化为特征向量的方法,并使用机器学习方法对未知功能序列进行功能标注一般步骤。
关键词:DNA序列;特征提取;功能预测;序列数据库
中图分类号:TP18 文献标识码:A 文章编号:1009-3044(2016)25-0151-02
1 引言
随着测序技术的迅速进步,各类生物数据库中的序列数据正在快速增长,生物大数据正在高速填充世界各生物公共平台的后台数据库;仅以美国国家生物技术信息中心(National Center for Biotechnology Information)的DNA序列数据库GenBank为例,截止2014年8月,GenBank数据库中的碱基对数量已超过13630亿对(Base Pair),较上一年增长了45%[1],该数据库的碱基对数量在2013年、2012年和2011年的年增长率分别是43%[2]、45.1%[3]和33.1%[4]。如此快速增长的序列数据,仅通过实验手段对这些序列数据进行功能注释显然已不现实,基于计算技术的快速功能注释已经变得势在必行。
DNA序列是由A,T,C和G四个字母组成的字符串,而目前的机器学习方法仅以特征向量作为输入;因此,将DNA序列转化为特征向量并尽可能保留序列内部的信息是特征提取技术的关键。
对新测序或者未知功能的DNA序列,对其功能进行验证的可靠方法是人工实验,但在数量庞大的DNA序列面前,全部由实验方法验证其功能显然已不可行,借助计算机领域的机器学习方法快速注释新序列的潜在功能便是一种可行的途径。这种功能注释方法的理论基础是序列的相似性意味着功能上的相似性。机器学习方法首先要获得一组DNA序列的训练集,该集合中的序列是已确定其功能的序列,由该训练集构建学习模型,并在训练集上进行交叉检验来验证该学习模型的预测性能,然后应用该模型对未知功能DNA序列进行功能预测。当然,并不是所有机器学习方法都适合对DNA序列的功能进行预测,因此,本文对DNA序列的特征向量提取方法及构建机器学习模型等问题进行了探讨。
2 DNA序列的特征提取策略
DNA序列由4种核苷酸碱基组成,分别是腺嘌呤(Adenine, A)、鸟嘌呤(Guanine,G)、胞嘧啶(Cytosine,C)和胸腺嘧啶(Thymine, T)。DNA序列的特征提取就是将由A、G、C和T四个字母组成的长串序列(字符串)转化成用数值表示的特征向量的过程。
基于k-mer的特征提取方法是一种常用方法。考虑由字母表∑={A,G,C,T}生成长度为k的序列片段(即k-mer),并统计这些片段在DNA序列中的出现频率,由这些频率值构造特征向量。当k=1时,即统计字母表∑中4个字母在序列中的出现频率,生成一个有4个分量的特征向量。当k=1时,一个特征向量仅有4个分量,一般没有意义。当k=2时,即计算集合∑2={AA, AG, AC, ..., TC, TT}中的16个双核苷酸碱基在DNA序列中的出现频率,由此构成一个有16个分量的特征向量。例如,一条DNA序列为“ACGT”,则该序列包含三个2-mer分别为AC、CG和GT,这三个2-mer的出现频率均为1/3=0.33;该序列生成一个有16个分量的向量,其中有三个分量为0.33,即为前面所提到的3个2-mer的出现频率。当k=3时,特征向量的长度为43,即64维。随着k的增大,特征向量的维度迅速升高,例如,当k=8时,表示这条DNA序列的特征向量长度为65536维(48=65536),如此高维的特征向量已引起维度灾难,机器学习算法在处理高维向量时,其性能会显著下降,k值并不是越高越好。
基于k-mer的特征提取方案,衍生出一系列的特征提取方法。比如,将不同k值的k-mer组合,生成混合特征向量。例如将k=1、k=2和k=3三类特征向量进行组合,生成具有84个分量(41+42+43=84)的特征向量。基于k-mer的编码思想,王树林[5]等人提出了基于k-mer的哈希编码方案。在他们的论文[5]中,将字母表∑中4个字母进行二进制编码:Code(A)=(00)2,Code(G)=(01)2,Code(C)=(10)2和Code(T)=(11)2,括号外的下标2表示二进制,编码函数Code(si)表示对字母表∑中的单字符si进行二进制编码,并将k-mer短序列通过哈希函数映射为离散的数值向量,其哈希函数f:∑k→N定义为:
s[1..k]表示长度为k的DNA短序列片段,即k-mer。例如,DNA序列为“ACGT”,当k=2时,2-mer“AC”的哈希映射可以表示为:f(AC)=42-1·Code(A) + 42-2·Code(C),计算得f(AC)=2;2-mer“CG” 可表示为f(CG)=42-1·Code(C) + 42-2·Code(G),计算得f(CG)=9;同理2-mer“GT” 可表示为f(GT)=42-1·Code(G) + 42-2·Code(T),计算f(GT)=7。因此,DNA序列“ACGT”可表示为向量(2,9,7)。
由于DNA序列由双链构成,字母表∑中的4个字符在双链上以互补配对方式出现,即A与T配对,C与G配对。为了消除双链中的单链特异性,Noble[6]和刘滨[7]等人应用反向互补k-mer对DNA序列进行向量化。比如,当k=2时,基本的2-mer有16个,即AA, AC, AG, AT, CA, CC, CG, CT, GA, GC, GG, GT, TA,TC, TG, TT,而考虑反向互补后的2-mer则缩减为10个,即为AA,AC, AG, AT, CA, CC, ‘CG, GA, GC, TA,生成的特征向量为10维。
与基于k-mer的特征提取方法不同,基于伪核苷酸组成成分[8](Pseudo dinucleotide composition)的特征向量提取方法不仅考虑了DNA序列中的局部顺序关系(即k-mer),也考虑了碱基的全局次序模式(即伪核苷酸组成)。例如,基于双核苷酸的伪双核苷酸组成的特征向量提取方法可以参考[9]。
3 常用DNA序列的分类算法
当使用合适的特征提取方法将DNA序列转化为特征向量后,便可以使用正负样本集合生成训练集。生物信息学领域应用比较广泛的机器学习算法包括支持向量机(Support Vector Machine, SVM)算法、随机森林(Random Forests, RF)算法和人工神经网络(Artificial Neutral Networks, ANN)等算法。
使用机器学习分类算法的流程一般包括,构建已知功能DNA序列的训练集,训练集包括正样本和负样本。正样本是指具有确定功能的DNA序列,负样本指不具有该功能的一般DNA序列。训练集中正负样本数量(DNA序列)应该保存一致。为了提高学习模型的预测性能,需要对训练集中的DNA序列通过去重复来消除偏倚,可使用blast[10]或cd-hit[11]等软件消除正负训练集中相似度较高的序列,正负训练集之间不能有重复序列,即交集为空,负训练集应该具有一般序列的代表性,然后应用特征提取方法将DNA正负样本序列转化为带有标号的特征向量集合,并输入到指定的机器学习算法构建预测模型。为了评价所构建模型的分类性能,一般在训练集上使用交叉检验来对模型进行评测。留一法[12] 交叉检验(leave one out cross validation)和5倍交叉检验[13](5-fold cross validation)都是比较常用的方法。5倍交叉检验是将训练集均分成5份,依次选其中的4份用作模型训练,用剩余一份用作测试集,如此重复5次;留一法则是选取训练集中的一个样本用作测试样本,剩余样本作为训练样本,依次让每个样本用作测试集仅一次,并用几项评测指标对分类模型进行评价。评测指标包括:灵敏度(Sensitivity, Sn)、特异度(Specificity, Sp)、准确度(Precision, Pr)、马修相关系数(Mathews correlation coefficient, MCC)等。
公式中的TP表示真阳性(True Positive),表示训练集正样本中被预测为正的样本数;TN表示真阴性(True Negative),即负样本中预测为负的样本数;FP表示假阳性(False Positive),表示训练集中负样本被预测为正的样本数;FN表示假阴性(False Negative),即正样本中被预测为负的样本数。通过上述指标,我们可以评价一个机器学习分类模型的性能,并通过修正机器学习算法中的参数使分类器的性能达到最优。
4 结束语
本文简单探讨了应用机器学习方法对DNA序列进行功能预测的方法,其步骤包括构建具有特定功能的DNA序列训练集,训练集中包括正样本和负样本;将正负样本通过DNA特征提取方法转化为特征向量集,然后应用一种机器学习算法对特征向量集进行训练,生成预测模型,使用交叉检验方法对预测模型进行参数调优,应用该模型即可对未知功能的DNA序列进行功能预测,判断未知功能DNA序列是否具有相应的功能。在本文中,我们简单介绍了使用机器学习算法对DNA序列进行功能预测的一般过程,希望能对机器学习方法在生物信息学领域的应用起到抛砖引玉的作用。
参考文献:
[1] K. Clark, I. Karsch-Mizrachi, D. J. Lipman, et al., "GenBank," Nucleic Acids Res, 2016(44):67-72.
[2] D. A. Benson, K. Clark, I. et al., "GenBank," Nucleic Acids Res, 2015(43):30.
[3] D. A. Benson, K. Clark, I. Karsch-Mizrachi, et al., "GenBank," Nucleic Acids Res,2014(42):32.
[4] D. A. Benson, M. Cavanaugh, K. Clark, I. Karsch-Mizrachi, D. J. Lipman, J. Ostell, et al., "GenBank," Nucleic Acids Res,2013(41):36-42.
[5] 王树林, 王戟, 陈火旺, 等.k-长DNA子序列计数算法研究[J].计算机工程,2007(33):3.
[6] W. S. Noble, S. Kuehn, R. Thurman, et al., "Predicting the in vivo signature of human gene regulatory sequences," Bioinformatics, 2005,21(1):338.
[7] B. Liu, R. Long, K. C. Chou, "iDHS-EL: identifying DNase I hypersensitive sites by fusing three different modes of pseudo nucleotide composition into an ensemble learning framework," Bioinformatics, Apr 8 2016.
[8] K. C. Chou, "Prediction of protein cellular attributes using pseudo-amino acid composition," Proteins,2001(43):246-55.
[9] W. Chen, T. Y. Lei, D. C. Jin, et al., "PseKNC: a flexible web server for generating pseudo K-tuple nucleotide composition," Anal Biochem,2014(456):53-60.
[10] S. F. Altschul, W. Gish, W. Miller, E. et al., "Basic local alignment search tool," J Mol Biol, 1990(215):403-10.
[11] W. Li,A. Godzik, "Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences," Bioinformatics, 2006(22):1658.
[12] H. Bhaskar, D. C. Hoyle, S. Singh, "Machine learning in bioinformatics: a brief survey and recommendations for practitioners," Comput Biol Med, 2006(36):1104-25.
[13] G. Liu, "Using weighted features to predict recombination hotspots in Saccharomyces cerevisiae," Journal of Theoretical Biology, 2016.
猜你喜欢
电子制作(2019年15期)2019-08-27
电子制作(2018年19期)2018-11-14
自动化学报(2017年11期)2017-04-04
中国生物医学工程学报(2017年6期)2017-02-10
广西科技大学学报(2016年1期)2016-06-22
计算机工程(2015年4期)2015-07-05
制造技术与机床(2015年10期)2015-04-09
机电信息(2015年3期)2015-02-27
机械工程师(2015年10期)2015-02-02
噪声与振动控制(2015年4期)2015-01-01
