
铱羰基碘配合物的制备及羰基基团变化研究
2016-11-29 07:43高山林李媛赖春波
上海化工 2016年3期
高山林 李媛 赖春波
上海华谊集团技术研究院(上海 200241)
铱羰基碘配合物的制备及羰基基团变化研究
高山林李媛赖春波
上海华谊集团技术研究院(上海200241)
铱羰基碘配合物是目前工业上应用羰基合成法生产醋酸的主催化剂,对羰基变化过程的研究有利于加强对催化剂失活过程的认识。采用13C NMR(核磁共振)等手段对制备的不同结构的铱羰基碘配合物进行表征。通过制备13CO标记的铱羰基碘配合物,证实在高水质量分数的醋酸体系中主要以活性中心cis-[Ir(13CO)2I2]-的形式存在,在无13CO氛围保护的加热条件下易氧化并失去13CO,推测形成[Ir(13CO)I4(H2O)]-和[Ir(13CO)I4]22-;在低水质量分数的醋酸体系中主要以[HIr(13CO)2I3]-和cis-[Ir(13CO)2I4]-的形式存在,在无13CO氛围保护的加热条件下失去13CO,推测形成[Ir(13CO)I4]22-及其同分异构体。
羰基合成铱羰基碘配合物13CNMR13CO标记
0 前言
甲醇低压羰基合成法是当前生产醋酸的最主要方法,据统计,目前全世界90%以上的醋酸采用该方法生产[1-5]。羰基合成可以高效地利用碳资源,减少碳排放,是发展绿色C1化工的典范。在羰基合成醋酸的生产工艺中,其主催化剂为铑或铱的羰基碘配合物,对于铑或铱作为主催化剂的羰基合成醋酸的催化循环机理已有详细的报道[6-10]。在二氯甲烷等非醋酸体系中进行铱羰基碘配合物的制备和表征也已有文献研究报道[11-17],但对在醋酸和水体系中进行铱羰基碘的催化剂制备并进行催化剂表征的报道较少,也未见利用核磁碳谱分析手段对铱羰基碘配合物在醋酸和水体系中的羰基变化进行研究的报道。在实际的工业生产中,不同的水质量分数对主催化剂的性能有明显影响,如催化剂在低CO分压下容易失活沉淀,形成的固体物沉积在闪蒸器底盘、破涡器、管道、换热器、泵滤网等地方,会产生堵塞作用,造成工业装置生产过程中紧急停车[18-23]。因此,本文在醋酸和水的体系中制备出13CO标记的铱羰基碘配合物,对其进行结构表征,并通过13C NMR(核磁共振)等手段研究羰基的变化过程。本研究有利于加深对现有甲醇低压羰基合成醋酸的催化剂的认识,有利于设计和改进现有催化体系,使醋酸生产工艺更加绿色高效。
1 实验部分
1.1实验原料
醋酸(质量分数为99.85%,工业优等品),上海华谊能源化工有限公司;普通CO气体(体积分数为99.9%),上海神开气体技术有限公司;13CO(体积分数为99%),美国西格玛奥德里奇(Sigma-Aldrich)公司;碘化锂(质量分数为99.9%),上海欧金实业有限公司;醋酸铱溶液(铱金属质量分数为4.09%),德国尤美科(Umicore)公司。
1.2实验方法
铱羰基碘配合物催化剂的制备:在200 mL锆材反应釜中,以一定量的水、醋酸、碘化锂为起始反应物,向其中添加含铱的催化剂前体,反应体系用CO(或13CO)置换3次,通入CO(或13CO)至压力为0.25 MPa;开启搅拌装置,并维持搅拌速率约为860 r/min,升温到120℃,通入CO(或13CO)至压力为0.5 MPa;维持反应温度为120℃,开始反应制备铱羰基碘配合物,间歇通入CO(或13CO),直至溶液不再吸收CO(或13CO)、反应釜压力不变为止;激冷,释放残余的CO(或13CO),并用低压氮气置换残余的CO(或13CO),取样进行分析。催化剂的制备实验装置如图1所示。
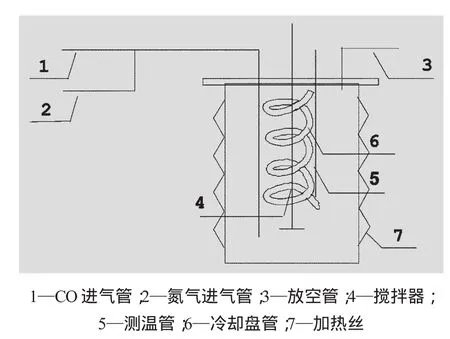
图1 催化剂制备装置
催化剂羰基变化跟踪实验:将反应制备的金属羰基碘配合物的溶液移入多通道的平行反应器中,以氮气置换出游离CO,并保持微正压;模拟工业装置苛刻的闪蒸条件,在135℃下,采用回流搅拌的运行方式进行催化剂羰基变化跟踪实验,实验结束后停止加热和搅拌,冷却后取样进行分析。
1.3分析仪器及方法
铱的质量分数通过Spectro Genesis电感耦合等离子发射光谱仪(ICP)进行分析,分析条件为:等离子气体流速为15 L/min,辅助气体流速为0.2 L/min,载气流速为0.8 L/min,功率为1300 W,进样流速为1.5 mL/min。制备的铱羰基碘配合物的核磁共振谱在Bruker AVANCE-400核磁共振仪上进行13C NMR的测定,常规CO配位的金属羰基碘配合物扫描10 240次,13CO标记的金属羰基碘配合物扫描128次。采用Thermo Finnigan公司的TSQ QuantumTMAccess电喷雾离子化三重四极杆质谱仪以负离子模式检测相对分子质量。在Nexus380傅里叶变换红外光谱仪上扫描红外谱图,采用傅里叶变换透射法,扫描32次,分辨率为2。
2 结果与讨论
2.1铱羰基碘配合物在醋酸水溶液中制备和表征
将铱催化剂前体和碘化锂溶于醋酸和水的混合溶液中,完全溶解后铱的质量分数约为5×10-3,在一定条件下通入CO(或13CO)气体,保证足够长的反应时间,直至溶液不再吸收CO(或13CO)为止。制备结束后的铱催化剂反应液通过ICP测定金属离子质量分数,通过13C NMR测定金属羰基碘化合物的特征羰基峰,分析结果如表1所示。
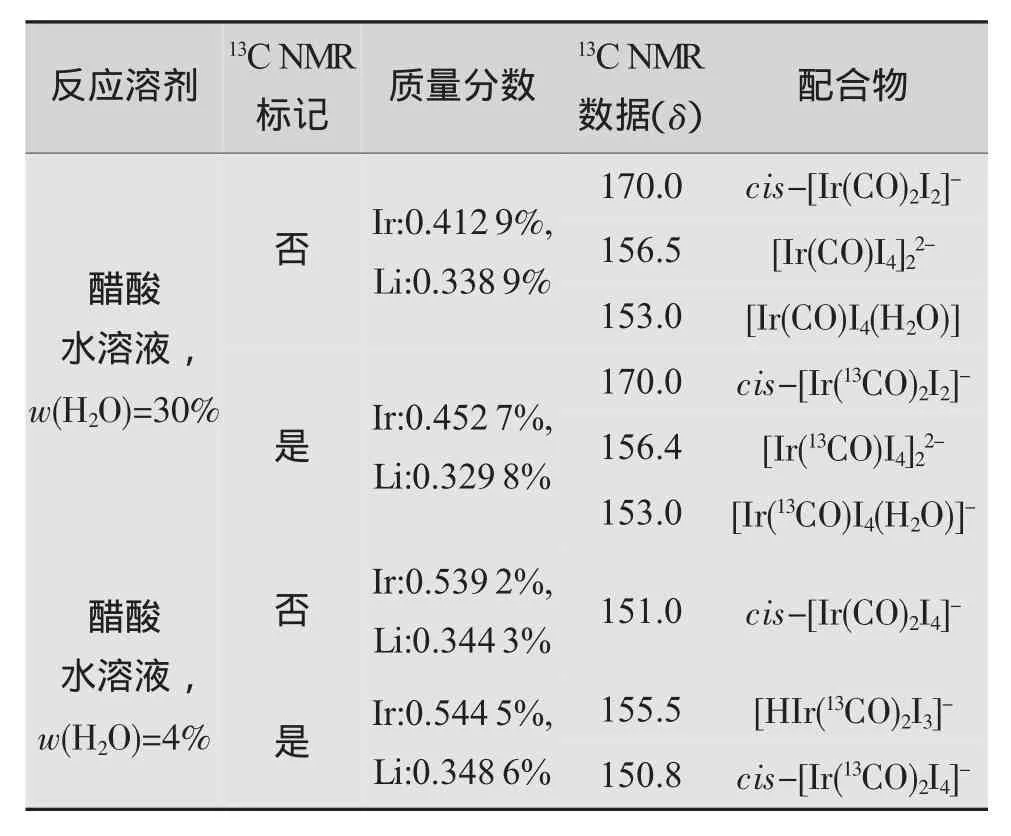
表1 铱羰基碘配合物的制备和表征
表1显示,在高水质量分数(30%,下同)条件下,制得的是+1价铱化合物cis-[Ir(CO)2I2]-[11-12,17],13C NMR分析结果表明,用常规CO制备得到的物种为+1价的催化剂活性中心[Ir(CO)2I2]-,质谱检测到[Ir(CO)2I2]-含同位素的离子信号:m/z=502.4,m/z= 500.2。室温放置一段时间后,13C NMR出峰δ=156.4,δ=153.0,1H NMR检测不到Ir—H的信号,质谱检测到含同位素的离子信号:m/z=728.0,m/z=726.0,对应红外出峰v(CO)=2 049 cm-1,推测生成了[Ir(CO)I4]22-或[Ir(CO)I4(H2O)]-[15]。在低水质量分数(4%,下同)的条件下制备的铱羰基碘配合物,放置一段时间=进行NMR分析检测得到的物种是cis-[Ir(CO)2I4]-[11,14-15],质谱检测到[Ir(CO)2I4]-含同位素的离子信号:m/z= 756.2,m/z=754.2,对应的红外出峰v(CO)=2116 cm-1,2 072 cm-1。同样制备了13CO标记的铱羰基碘配合物,高水质量分数条件下制备得到cis-[Ir(13CO)2I2]-,质谱检测到[Ir(13CO)2I2]-含同位素的离子信号:m/z= 504.5,m/z=502.4;随着放置时间的延长,逐渐形成了[Ir(13CO)I4]22-或[Ir(13CO)I4(H2O)]-,质谱检测到含同位素的离子信号:m/z=729.1,m/z=727.0,对应的红外出峰v(13CO)=2000 cm-1。在低水质量分数条件下,除了得到cis-[Ir(13CO)2I4]-,质谱检测到含同位素离子信号:m/z=758.1,m/z=756.1,对应红外出峰v(13CO)=2 067 cm-1,2 026 cm-1;还观察到生成了[HIr(13CO)2I3]-[11,14]:13C NMR δ=155.5,1H NMR δ=-11.1,2JH-13CO=5.4 Hz,对应红外出峰v(Ir—H)=2157(w)cm-1。
2.213CO标记铱催化剂羰基变化过程跟踪和分析
通过模拟工业装置闪蒸的实验评价方法,跟踪研究13CO标记铱羰基碘配合物的羰基变化过程。在高水质量分数体系下制备的铱羰基碘配合物,在室温下放置,通过13C NMR跟踪其中的羰基峰,发现刚制备的配合物由于13CO与cis-[Ir(13CO)2I2]-(δ=170.0)存在快速交换,因而在δ=170.0处出现宽峰[13],且有非常微弱的cis-[Ir(13CO)2I4]-(δ=150.1)峰;随着时间的延长,游离的13CO消失,cis-[Ir(13CO)2I2]-的峰变窄,其被氧化并进一步失去13CO,推测形成+3价铱[Ir(13CO)I4]22-(δ=156.5)及与水络合的Ir[(13CO)I4(H2O)]-(δ=153.0);室温放置1个月,随着时间的延长,+1价铱化合物的峰明显减弱,而对应+3价铱化合物的峰明显增强。13C NMR的跟踪分析见图2。
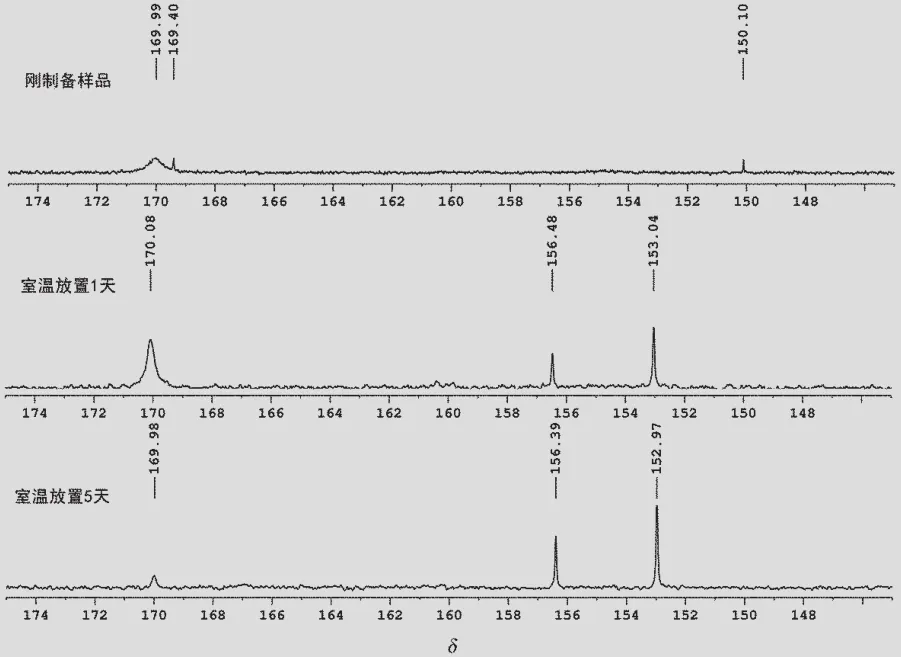
图2 高水质量分数条件下制备的13CO标记铱配合物在室温下的变化情况
而在低水质量分数体系中制备的铱羰基碘配合物,通过13C NMR跟踪羰基峰,发现制备后物种主要是[HIr(13CO)2I3]-(δ=155.5)和cis-[Ir(13CO)2I4]-(δ= 150.8),以及极少量的trans-[Ir(13CO)2I4]-[14-15](δ= 163.2)物质和[Ir(13CO)I4]22-(δ=157.0)物质。配合物[HIr(13CO)2I3]-不稳定,与HI生成cis-[Ir(13CO)2I4]-;室温放置1个星期后,[HIr(13CO)2I3]-完全消失,而且极少量的trans-[Ir(13CO)2I4]-也完全消失了,最后剩下的物种是cis-[Ir(13CO)2I4]-和少量的[Ir(13CO)I4]22-。通过13C NMR的跟踪分析,结果见图3。
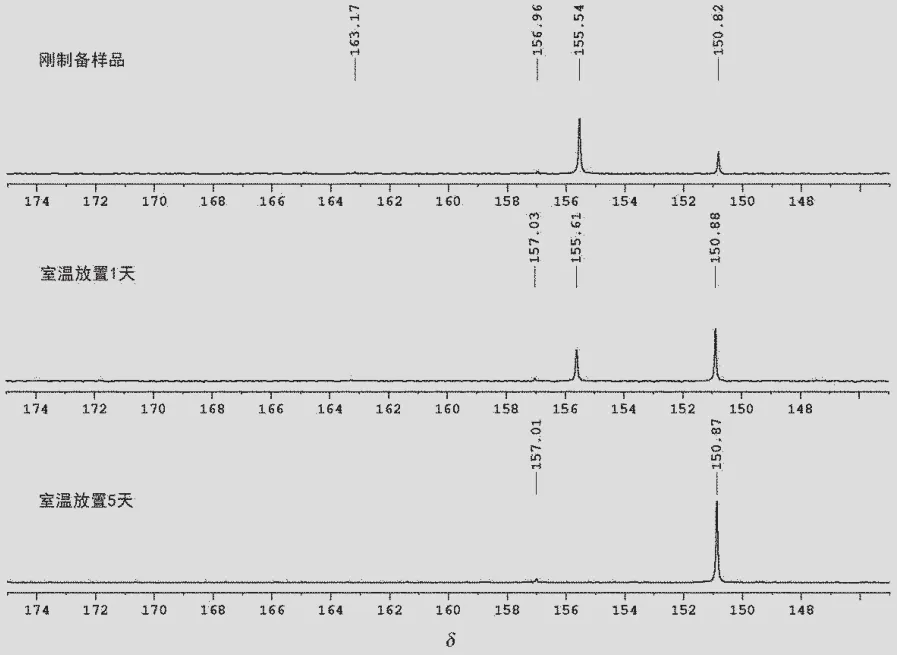
图3 低水质量分数条件下制备的13CO标记铱配合物在室温下的变化情况
模拟工业生产装置的闪蒸条件,用氮气置换出游离的13CO,在加热实验前分析铱羰基碘配合物样品,在高水质量分数条件下制备的铱羰基碘配合物主要为+1价活性中心[Ir(13CO)2I2]2-(δ=170.1),并且经过推测而得出的+3价铱物种[Ir(13CO)I4]22-(δ= 156.5)和[Ir(13CO)I4(H2O)]-(δ=153.0)。在135℃下加热,1.5 h后,+1价[Ir(13CO)2I2]2-的峰逐渐减弱,在δ= 160.4,δ=158.1处出现新峰。加热3.0 h后,其他物种的羰基峰消失,只剩[Ir(13CO)I4]22-和[Ir(13CO)I4(H2O)]-的核磁峰。经加热实验后,反应液的红外谱图只出现单峰v(13CO)=2000 cm-1。13C NMR跟踪分析见图4。
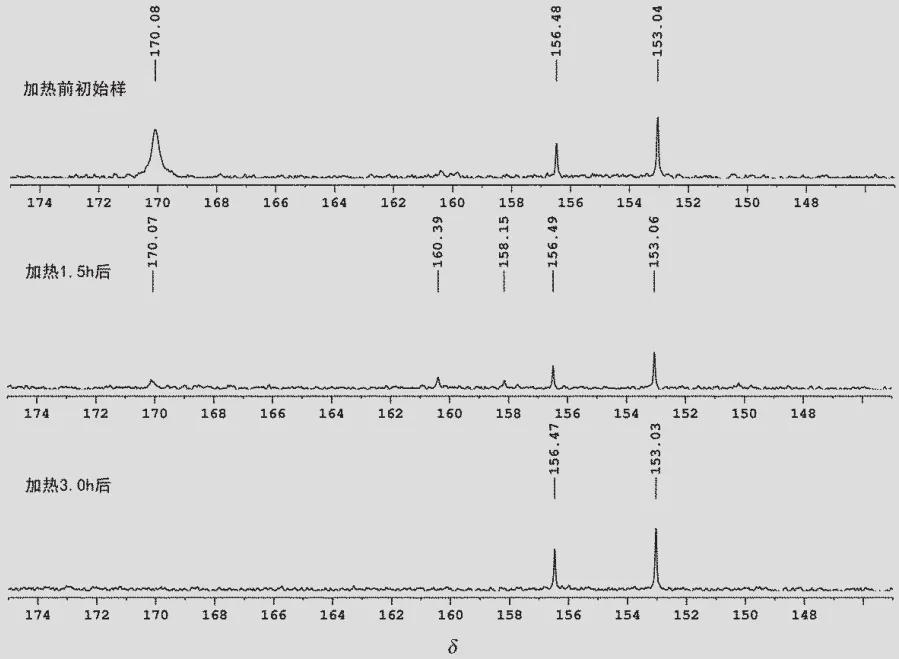
图4 高水质量分数条件下制备的13CO标记铱配合物在135℃下加热时的变化情况
同样模拟闪蒸条件,用氮气置换出13CO,在实验之前对低水质量分数条件下制备的铱羰基碘配合物样品进行了分析,主要为[HIr(13CO)2I3]-(δ=155.5)和cis-[Ir(13CO)2I4]-,极少量trans-[Ir(13CO)2I4]-(δ=163.2)和[Ir(13CO)I4]22-(δ=157.0)。加热1.5 h,[HIr(13CO)2I3]-的峰减弱,cis-[Ir(13CO)2I4]-的峰增强,在δ=153.6处出现小峰,推测生成[Ir(13CO)I4(H2O)]-;在δ=157.0和δ= 157.1处也出现新峰,推测生成[Ir(13CO)I4]22-和其化合物的同分异构体。加热3.0 h后,trans-[Ir(13CO)2I4]-的峰完全消失,而且发现[HIr(13CO)2I3]-的峰进一步减弱,cis-[Ir(13CO)2I4]-仍然大量稳定地存在,反应液最终的红外谱图显示,13CO的吸收峰位于2 068,2 027和1992 cm-1处。13C NMR的过程分析见图5。
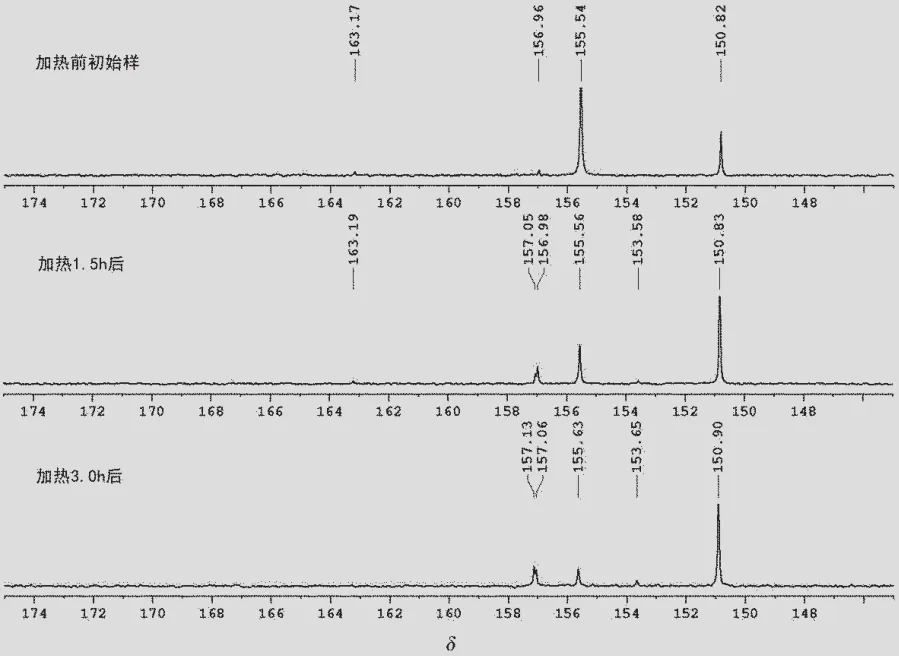
图5 低水质量分数条件下制备的13CO标记铱配合物在135℃下加热时的变化情况
模拟工业装置苛刻的闪蒸条件进行实验之后,质谱分析只检测到[Ir(13CO)I4]-、[Ir(13CO)2I4]-的特征峰,未检测到[IrI4]-的特征峰,铱催化剂反应液澄清,无明显沉淀。
对铱催化剂羰基变化过程的推测见图6,+1价铱催化剂的活性中心cis-[Ir(13CO)2I2]-易与HI发生氧化加成反应,生成+3价的铱配合物[HIr(13CO)2I3]-,该配合物进一步与HI作用生成cis-[Ir(13CO)2I4]-,cis-[Ir(13CO)2I4]-易失去一分子13CO;当水质量分数高时,推测其更易形成[Ir(13CO)I4(H2O)]-和[Ir(13CO)I4]22-;而当水的质量分数低时,更易形成[Ir(13CO)I4]22-及其同分异构体。与文献报道的铑羰基碘配合物相比,铱羰基碘配合物的羰基结合更稳定,因而催化剂更加稳定。
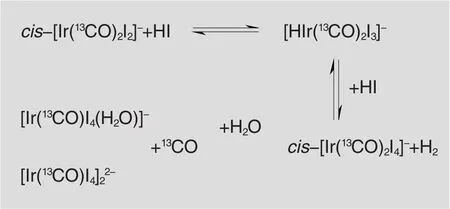
图6 推测的铱催化剂羰基变化过程
3 结论
通过制备13CO标记的铱羰基碘配合物,证实了其在高水质量分数的醋酸体系中主要以活性中心cis-[Ir(13CO)2I2]-的形式存在,在无13CO氛围保护、室温或工业闪蒸条件下加热时易被氧化并失去13CO形成+3价的铱配合物,推测其结构为[Ir(13CO)I4]22-和[Ir(13CO)I4(H2O)]-;在低水质量分数的醋酸体系中主要以[HIr(13CO)2I3]-和cis-[Ir(13CO)2I4]-的形式存在,[HIr(13CO)2I3]-室温下会逐渐转化成cis-[Ir(13CO)2I4]-,但在工业闪蒸条件加热下易失去13CO,推测主要生成[Ir(13CO)I4]22-及其同分异构体。13CO的失去是导致催化剂失活或沉淀的主要原因之一,相对于铑催化剂而言,铱催化剂更为稳定。
[1]Maitlis P M,Haynes A,SunleyG J,et al.Methanol carbonylation revisited:thirty years on[J].Journal of the Chemical Society,Dalton Transactions,1996(11):2187-2196.
[2]王玉和,贺德华,徐柏庆.甲醇羰基化制乙酸[J].化学进展,2003,15(3):215-221.
[3]郑修成,张守民,黄唯平,等.甲醇羰基化制醋酸铱基催化剂体系的研究[J].有机化学,2003,23(6):613-618.
[4]曹宏兵.铱催化剂催化甲醇羰基化制备乙酸研究进展[J].天然气化工:C1化学与化工,2011,36(6):46-52.
[5]周斌,孙大贵,杜军,等.甲醇低压羰基法合成醋酸的催化剂体系[J].化学通报,2010(12):1093-1098.
[6]Sarmah P P,Dutta D K.Rhodium(Ⅰ)carbonyl complexes of quinoline carboxylic acid:synthesis,reactivity and catalytic carbonylation reaction[J].Journal of Molecular Catalysis A:Chemical,2013,372:1-5.
[7]Dutta DK,Deb B,Hua GX,et al.Chelate and trans effect of P,O donor phosphine ligands on rhodium catalyzed carbonylation of methanol[J].Journal of Molecular Catalysis A:Chemical,2012,353-354:7-12.
[8]Borah B J,Deb B,Sarmah P P,et al.Synthesis,reactivities and catalytic carbonylation of rhodium(Ⅰ)carbonyl complexes containing isomeric acetylpyridine ligands[J].Inorganica Chimica Acta,2011,370(1):117-121.
[9]Elliott P I P,Haak S,Meifer A J H M,et al.Reactivity of Ir (III)carbonyl complexes with water:alternative by-product formation pathways in catalytic methanol carbonylation[J]. Dalton Transactions,2013,42(47):16538-16546.
[10]Dutta D K,Deb B.Potential rhodium and ruthenium carbonyl complexes ofphosphine-chalcogen(P-O/S/Se)donor ligands and catalytic applications[J].Coordination Chemistry Reviews,2011,255:1686-1712.
[11]Whyman R,Wright A P,Iggo J A,et al.Carbon monoxide activation in homogeneously catalysed reactions:the nature and roles of catalytic promoters[J].Journal of the Chemical Society,Dalton Transactions,2002(5):771-777.
[12]Gautron S,Giordano R,Le Berre C,et al.Isolation and structural characterization of anionic and neutral compoundsresulting from the oxidative addition of HI or CH3I to[IrI2(CO)2]-[J].Inorganic Chemistry,2003,42(18):5523-5530.
[13]Churlaud R,Frey U,Metz F,et al.Mechanism of CO exchange on cis-[M(CO)2X2]-complexes(M=Rh,Ir;X=Cl,Br, or I)[J].Inorganic Chemistry,2000,39(2):304-307.
[14]Gautron S,Lassauque N,Le Berre C,et al.Promoting role of [PtI2(CO)]2in the iridium-catalyzed methanol carbonylation to acetic acid and its interaction with involved iridium species[J].Organometallics,2006,25(25):5894-5905.
[15]Haynes A,Mcnish J,Pearson J M.Cis-trans isomerism in [M(CO)2I4]-(M=Rh,Ir):kinetic,mechanistic and spectroscopic studies[J].Journal of Organometallic Chemistry, 1998,551(S1-2):339-347.
[16]Malatesta L,Naldini L,Cariati F.Iodocarbonyl derivatives of iridium[J].Journal of the Chemical Society(Resumed),1964:961-965.
[17]Haynes A,Maitlis P M,Morris G E,et al.Promotion ofiridium-catalyzed methanol carbonylation:mechanistic studies of the cativa process[J].Journal of the American Chemical Society,2004,126(9):2847-2861.
[18]Le Berre C M,Nguyen D H,Serp P G,et al.Production of acetic acid with enhanced catalyst stability:US,13/715168 [P].2012-12-14.
[19]Torrence P G.Acetic acid production methods incorporating tin or ruthenium catalyst stabilizers:EP,20060734757[P]. 2006-02-10.
[20]赖春波,王苏,高山林,等.一种羰化合成醋酸的生产方法:中国,201110300797.3[P].2011-09-28.
[21]高山林,赖春波,王苏,等.一种羰化合成醋酸的生产方法:中国,201210271420.4[P].2012-08-01.
[22]王苏,赖春波,白云飞,等.适用于C1-C4醇及其衍生物羰基化反应的催化剂:中国,201210558033.9[P]. 2012-12-20.
[23]高山林,赖春波,王苏,等.通过催化羰基化反应制备乙酸的方法:中国,201210558083.7[P].2012-12-20.
O643.36
上海市优秀技术带头人计划项目(15XD1521700)
高山林男1982年生硕士工程师主要从事羰基化反应研究
2015年12月
猜你喜欢
氮肥与合成气(2023年2期)2023-02-14
云南化工(2021年11期)2022-01-12
云南化工(2021年7期)2021-12-21
陶瓷学报(2021年5期)2021-11-22
广州化工(2020年8期)2020-05-19
世界有色金属(2018年8期)2018-06-28
化工学报(2016年10期)2016-10-13
华东理工大学学报(自然科学版)(2015年5期)2015-02-27
应用化工(2014年1期)2014-08-16
无机化学学报(2014年6期)2014-02-28
