
对乙酰氨基酚-水复合物中氢键作用的理论研究
2016-12-14 06:04李玉莹王晓红黄正国
天津师范大学学报(自然科学版) 2016年4期
原 媛,孙 乐,李玉莹,王晓红,黄正国
(天津师范大学 a.化学学院,b.无机-有机杂化功能材料化学教育部重点实验室,c.天津市功能分子结构与性能重点实验室,天津 300387)
对乙酰氨基酚-水复合物中氢键作用的理论研究
原 媛,孙 乐,李玉莹,王晓红,黄正国
(天津师范大学 a.化学学院,b.无机-有机杂化功能材料化学教育部重点实验室,c.天津市功能分子结构与性能重点实验室,天津 300387)
为了解对乙酰氨基酚(AP)在水溶液中的构象,在MP2/6-311++G(d,p)水平上研究了AP与水分子通过不同种氢键作用形成的6种AP-H2O复合物.首先分析这些复合物的几何结构、能量和振动频率,然后运用分子中原子的量子理论(QTAIM)、自然键轨道理论(NBO)和定域分子轨道-能量分解分析(LMO-EDA)等理论和方法对AP-H2O复合物中的氢键相互作用进行定性和定量分析.研究结果表明:(1)AP分子中的—CH基团与羰基氧原子之间形成的C8H8A···O2A分子内氢键在多数AP-H2O复合物中被保留下来;(2)AW1和AW6中的分子间氢键强于其他氢键;(3)AP的羰基氧原子是最有可能与H2O分子形成氢键的位点,第2个最有可能与H2O分子形成氢键的位点是AP的酚羟基,前者中H2O是质子供体,而后者中H2O则是质子受体;(4)AP-H2O的形成过程中,氢键作用与结构畸变都会在一定程度上决定AP-H2O复合物的相对稳定性,但前者的作用更大.
对乙酰氨基酚;复合物;氢键作用;自然键轨道理论(NBO);分子中原子的量子理论(QTAIM)
对乙酰氨基酚(acetaminophen,AP)的分子式为C8H9NO2,通常为白色结晶粉末.由于具有解热镇痛的作用,因此临床上常被用于感冒发烧、关节痛、神经痛、偏头痛、癌痛的缓解以及术后止痛等方面[1].AP作为止痛剂,具有较低的肾毒性[2].如AP和布洛芬都可以缓解成人骨关节炎的慢性疼痛[3],如果按照推荐剂量用药,AP的副作用相对于布洛芬更低[4].目前关于AP分子结构的研究较少,多采用红外和拉曼光谱技术进行.如Beames等[5]研究了喷射冷却的AP的共振双光子离子光谱,根据理论计算分析了其基态的构型和振动频率.Danten等[6]研究了AP分别和乙醇和丙酮形成的复合物.Lee等[7]在超分子束中发现了AP的2个同分异构体,并通过UV-UV烧孔光谱区分出二者为正反异构.
由于AP在临床上应用广泛,因此有必要通过研究其在水溶液中的构象来了解其药理学活性.表征药物或生物体系的构象时,通常弱相互作用尤其是氢键作用非常重要.为此本研究在计算了AP和H2O单体构成的AP-H2O复合物的能量、结构和简谐振动频率的基础上,运用分子中原子的量子理论(QTAIM)[8-10]和自然键轨道理论(NBO)[11-12],结合定域分子轨道分解分析法(LMO-EDA)[13],深入探讨了AP-H2O复合物的氢键本质.
1 计算细节
首先,采用MP2方法结合6-311++G(d,p)基组分别对AP和H2O单体的构型进行全优化,在此基础上构建AP-H2O复合物,并在同一计算水平上对所有复合物进行全优化.通过简谐振动频率计算可以获得APH2O复合物的零点振动能(ZPVE),并确保获得的复合物具有能量极小值.考虑到基组重叠误差(basis set superposition error,BSSE)的影响,采用均衡校正方法对优化后的结构进行BSSE校正.在ZPVE和BSSE修正的基础上进行相互作用能计算.最后,通过NBO、QTAIM和LMO-EDA等方法分析AP-H2O复合物中氢键相互作用的性质.用Gaussian09程序完成NBO分析[14],在MP2/6-311++G(d,p)水平上用AIM2000[15]软件获得的波动函数来进行QTAIM分析,在相同水平上采用Gamess程序完成LMO-EDA分析[16-17].
2 结果与分析
在MP2/6-311++G(d,p)水平上优化的AP和H2O单体的分子结构如图1所示.由图1可以看出,H2O中的羟基和氧原子可以作为质子供体/受体形成氢键. AP中,质子供体主要是苯环中的亚氨基和酚羟基,在一些复合物中—CH基团也可作为质子供体;质子受体主要有羟基氧原子、亚氨基氮原子和羰基氧原子,而且羰基氧原子通常接受—CH基团中的质子形成分子内氢键.
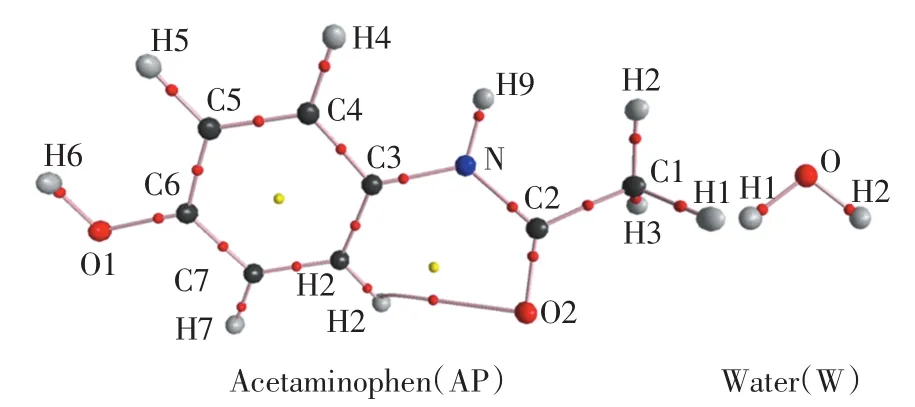
图1 AP与水分子单体的分子图Fig.1 Molecular graphs of acetaminophen(AP)and water(W)monomers
2.1 AP-H2O复合物的结构
优化后的AP-H2O复合物的分子结构如图2所示.

图2 AP-H2O复合物的分子图Fig.2 Molecular graphs of AP-H2O complexes
由图2可以看出,AP-H2O复合物的结构多样,共有6种类型.根据QTAIM理论,不论是分子间氢键还是分子内氢键,都可以用质子供体(X—H)和质子受体(Y)之间的键临界点(BCP)来表征;同时,多个氢键形
成的环结构中存在环临界点(RCP),RCP和相应的BCP之间的距离可以表征氢键结构的稳定性,距离越短则稳定性越弱[18-21].AP苯环中的环临界点与氢键的形成无关.AP单体中的—CH基团和氧原子之间形成了1个C8H8A…O2A分子内氢键(上标A代表对乙酰氨基酚),该氢键存在于复合物AW1、AW2、AW4和AW6中,并且这4种复合物中都有2个分子间氢键. AW1中,H2O单体中的氧原子同时接受来自AP单体的—CH基团和羟基提供的2个质子,形成分叉分子间氢键;AW2中的氢键形成类似于AW1;AW4和AW6中,H2O单体同时作为质子供体和质子受体与AP单体形成2个分子间氢键,分子间氢键OH1W…O2A和CHA…OW(上标中的W代表H2O)之间存在协同作用.与这4种AP-H2O复合物相比,AW3和AW5中发生了严重的结构形变,AW3有2个分子间氢键,而AW5只有1个分子间氢键.这是因为AW3中,AP单体的C8H8A…O2A分子内氢键被破坏后,H2O单体同时作为质子供体和质子受体与AP单体形成了2个分子间氢键;而AW5中的C8H8A…O2A分子内氢键被破坏后,H2O单体中的羟基向AP单体中的羰基氧原子提供质子形成了1个分子间氢键.
AP-H2O的6种复合物、AP单体以及H2O中氢键的结构参数如表1所示.
复合物中X—H键与自由单体(AP或H2O)中X—H键的差异可用来表征形成复合物后氢键的性质,X—H键伸长表明其为红移氢键,X—H键缩短表明其为蓝移氢键.X—H键或者H…Y改变的程度还可以反映氢键作用的强度.由表1可以看出,对于复合物中大多数以—CH基团作为质子供体而形成的氢键来说,其ΔRX—H值为负或者几乎不变,表明这些为很弱的蓝移氢键.而复合物中其他氢键的ΔRX—H为正值,表明其为红移氢键.AW6的OH1W…O2A分子间氢键ΔRX—H值最大,为0.001 0 nm,表明该氢键作用最强;其次是复合物AW1中的分子间氢键O1H6A…OW,该氢键作用也较强.ΔRX—H并不是判断氢键强度的唯一标准,可以用RH…Y来代替ΔRX—H判断氢键强弱.一般来说,H…Y越短,表明氢键作用越强.根据这一判断标准,AW1中的分子间氢键O1H6A…OW的RH…Y最小,因此强度大于AW6的分子间氢键OH1W…O2A.这种判断标准只适用于相似环境中种类相近的氢键.由于在很多情况下氢键的质子供体或受体类型不同,因此无法直接根据RH…Y判断氢键作用的强度.为此,本研究引入了氢键参数δRH…Y[22]来评价不同类型的成对原子之间的氢键强度,计算公式如下:

以氢键参数δRH…Y作为判断标准,AW1中分子间氢键的δRH…Y为0.084 2 nm,作用强度大于AW6中分子间氢键(0.083 5 nm)的强度,二者均为强氢键.在AW1、AW2、AW3、AW4和AW6中,以—CH基团作为质子供体与O原子形成的分子内氢键和分子间氢键,其δRH…Y均较小(<0.05 nm),表明这些氢键中—CH基团和Y原子的相互作用非常弱,二者之间存在部分范德华相互作用.另外,AW3和AW6中的分子间氢键OH1W…O2A的δRH…Y值大于AW5中的数值,这可能是由于前两者中的分子间氢键存在协同效应,使其强度增大,这种效应并未在其他的AP-H2O复合物中找到.

表1 在MP2/6-311++G(d,p)水平上计算的AP-H2O复合物中的氢键结构参数Tab.1 Structural parameters of H-bonds in AP-H2O complexes calculated at the MP2/6-311++G(d,p)level
2.2 氢键振动频率
在MP2/6-311++G(d,p)水平上计算AP-H2O复合物和单体中氢键的简谐振动频率及其变化值,结果如表2所示.

表2 AP-H2O复合物及其单体中氢键的伸缩振动频率(强度)Tab.2 Stretching vibrational frequencies(strength)of H-bonds both in AP-H2O complexes and monomers
对于常规的X—H…Y,氢键的形成会使X—H变弱,从而X—H伸缩振动频率发生红移,并且氢键越强其红移值越大,这也是X—H…Y的主要特征之一.如果X—H伸缩振动模与其他振动模混合,情况就会变得比较复杂.由表2结果可知,在AP单体中C1H1和H3—C2—H2间的对称和反对称伸缩振动模相混合,振动频率分别为3 086.2 cm-1和3 178.3 cm-1,因此对于以C1H1作为质子供体的氢键来说,就有2个ΔνX—H值.类似情况也出现在AP-H2O复合物中.如AW4中OH1W…O1A对称伸缩振动模混合了O1H6的伸缩振动模,根据二者各自在H2O单体中的伸缩振动模计算得到的ΔνX—H值分别为-33.2cm-1和-67.9cm-1.6种复合物的分子间氢键进行比较,AW6中OH1W…O2A的红移值最大,为-173.8 cm-1;AW1中的O1H6A…OW和AW3中的OH1W…O2A红移值略低,分别为-151.2 cm-1和-104.1 cm-1.这表明后两者的分子间氢键作用弱于AW6.AW4中的OH1W…O1A和AW5中的OH1W…O2A的红移值最低,即二者的氢键作用最弱.比较不同复合物的OH1W…O2A,AW6和AW3中的ΔνX—H显著大于AW5复合物中的数值,即后者的氢键作用小于前两者,这也证实了AW6和AW3中分子间氢键存在较强的协同效应.
2.3 QTAIM分析
QTAIM是研究氢键相互作用的常用方法[23-24],H…Y键之间临界点(BCP)的电子密度(ρb)及其Laplacian值(Δ2ρb)能够很好地衡量氢键强度.本研究中所有AP-H2O复合物和单体中氢键BCP上的电子密度拓扑性质如表3所示.

表3 AP-H2O复合物中H…Y氢键临界点处的电子密度拓扑性质Tab.3 Topological features of electronic density at H…Y BCPs of H-bonds in AP-H2O complexes
由表3可以看出,所有的复合物和AP单体中氢
键的Hb和Δ2ρb均为正值,且都落在Koch等[25]所提出的标准范围之内,因此这些氢键的强度均为微弱或中等,尤其是以—CH基团作为质子供体所形成的氢键,ρb和Δ2ρb的值都非常接近Popelier[10]所提出标准的下限,这表明它们是非常弱的氢键,具有部分色散作用的性质.比较OH1W…O2A分子间氢键在不同复合物中的强度,AW3和AW6中该氢键有较大的ρb和Δ2ρb值,显著高于AW5中的数值,表明前两者的氢键作用较强,验证了2.1中的结论.
2.4 NBO分析
对6种AP-H2O复合物进行NBO分析,结果如表4所示.

表4 AP-H2O复合物中H…Y氢键的二阶微扰能E(2)Tab.4 Second-order perturbation energies E(2)of H-bonds in AP-H2O complexes kJ/mol
由表4可知,在AW2的C8H8A…O2A分子内氢键和AW6的C1H1A…OW分子间氢键中,作为质子受体的氧原子只有1个sp分支,而其他复合物中作为质子受体的氧原子有2个分支:一个是sp杂化轨道,另一个是p杂化轨道,分别对应1个E(2)值.6种复合物中,AW1复合物中的O1H6A…OW分子间氢键有较大的E(2)值(47.52 kJ/mol),表明其发生了最强的电荷转移(CT)效应,这种效应对于氢键相互作用的贡献最大;其次是AW6中的OH1W…O2A分子间氢键,其E(2)值也较大,为42.51 kJ/mol,即CT效应在该分子间氢键中也起着重要作用.比较不同复合物中OH1W…O2A分子间氢键的强度,AW6和AW3中该氢键的E(2)值明显比AW5中的数值大,这也证实了前2种复合物中分子间氢键存在协同效应.以—CH基团作为质子供体的氢键的E(2)值小于3.5 kJ/mol,且远远小于其他氢键,表明这种氢键非常弱,与前面分析一致.对于QTAIM分析所证明的一些氢键,NBO分析无法找到,可能是由于这些氢键太弱,运用该方法检测不到.
2.5 LMO-EDA分析
为了探索氢键相互作用的本质,本研究在MP2水平上用LMO-EDA[13]方法进行了能量分解分析,结果如表5所示.

表5 在MP2水平上计算的AP-H2O复合物的LMO-EDA结果Tab.5 LMO-EDA results of AP-H2O complexes at MP2 level kJ/mol
由表5可以看出,复合物中AP单体和H2O之间的总相互作用能范围为-26.29~-15.72 kJ/mol.6种复合物中,AW6的相互作用能的绝对值最大,为-26.29 kJ/mol,因此AW6是最稳定的复合物,H2O分子最倾向与AP的羰基氧原子形成氢键.在AW6中,绝对值最大的稳定能是交换能(-56.18 kJ/mol);其次是静电能(-53.38 kJ/mol);AW6中又有强大的排斥能(100.53 kJ/mol);氢键的形成改变了单体片段原来的轨道形状,从而产生了-15.63 kJ/mol的极化能;相互作用能中最小的一部分是色散能,仅为-1.63 kJ/mol. AW1的相互作用能为-25.75 kJ/mol,稳定性仅次于AW6,其AP的酚羟基是第2个最有可能与H2O分子形成氢键的位点.不同于AW6中以羰基氧原子为质子受体与H2O形成分子间氢键,AW1中是以酚羟基作为质子供体与H2O形成分子间氢键.AW1中,对相互作用能贡献最大的也是静电能(-47.44 kJ/mol)和交换能(-49.12 kJ/mol),极化能(-14.04 kJ/mol)和色散能(-3.76 kJ/mol)对相互作用能的贡献都较小.AW3的稳定性较弱,相互作用能仅为-15.72 kJ/mol.
AP-H2O复合物中氢键的相互作用并不是影响其稳定性的唯一因素.根据NBO理论,AP-H2O复合物的稳定性还受到单体(AP和H2O)结构形变的影响,即形成复合物的过程中会产生变形能.由表5可以看出,所有复合物的变形能均小于2.2 kJ/mol,这预示着APH2O复合物都具有微小的结构形变,但对这些复合物稳定性的影响不大.
3 结论
本研究通过MP2方法结合6-311++G(d,p)基组分析了AP-H2O复合物的结构、能量和振动频率.除了复合物AW3和AW5之外,C8H8A…O2A分子内氢键在其他AP-H2O复合物中都被保留下来.AW3和AW6中的OH1W…O2A分子间氢键由于存在协同效应,氢键作用增强,这种协同效应并没有在其他复合物中发现.AW1中的O1H6A…OW分子间氢键和AW6中的OH1W…O2A分子间氢键都属于强氢键,AP-H2O复合物中以—CH基团作为质子供体的氢键属于弱氢键. AP的羰基氧原子是最有可能与H2O分子形成氢键的位点,而第2个最有可能与H2O分子形成氢键的位点是AP的酚羟基,前者中H2O是质子供体,而后者中H2O则是质子受体.氢键的相互作用和结构形变在复合物的相对稳定性中均起作用,但前者起主要作用. AW6的相互作用能最大,表明它是最稳定的复合物,其中分子间氢键(OH1W…O2A)的相互作用对复合物的稳定性贡献最大,而结构形变的影响较弱.
需要指出的是,由于AP中存在多个氢键形成位点,而本研究中涉及的仅是AP单体与水分子以分子数量比例为1∶1形成的AP-H2O复合物.随着H2O分子数目的增加,将会形成结构构象更为复杂的AP-H2O复合物.总之,H2O作为溶剂对AP的结构和性能的影响相当复杂,本研究仅是探讨AP微溶剂化的第一步,希望能够为进一步研究AP在水溶液中的结构提供重要信息.
[1]王孝文.非癌性疼痛药物治疗中对乙酰氨基酚的最新应用进展[J].中国新药杂志,2015,24(18):2085-2090. WANG X W.Advance in application of acetaminophen in the treatment of noncancer pain[J].Chinese Journal of New Drugs,2015,24(18):2085-2090(in Chinese).
[2]施文,王永铭,李端,等.对乙酰氨基酚与非甾体抗炎药治疗骨关节炎的疗效和安全性的观察[J].中国疼痛医学杂志,2004,10(6):327-331. SHIW,WANGYM,LID,etal.Theefficacyandsafetyofacetaminophen versus NSAIDs in osteoarthritis treatment[J].Chinese Journal of Pain Medicine,2004,10(6):327-331(in Chinese).
[3]HUGHES J.Pain Management:From Basics to Clinical Practice[M]. Edinburgh:Churchill Livingstone,2008.
[4]王生俊.扑热息痛的毒副作用与酒精[J].北方药学,2010,7(3):45-46. WANG S J.Toxic and side effects of paracetamol and alcohol[J].Journal of North Pharmacy,2010,7(3):45-46(in Chinese).
[5]BEAMES J M,HUDSON A J.Jet-cooled spectroscopy of paracetamol[J]. Phys Chem Chem Phys,2010,12(16):4157-4164.
[6]DANTEN Y,TASSAING T,BESNARD M.Density functional theory(DFT)calculations of the infrared absorption spectra of acetaminophen complexes formed with ethanol and acetone species[J].J Phys Chem A,2006,110(28):8986-9001.
[7]LEE S J,MIN A,KIM Y,et al.Conformationally resolved structures of jet-cooled acetaminophen by UV-UV hole-burning spectroscopy[J].Phys Chem Chem Phys,2011,13(37):16537-16541.
[8]BADER R F W.Atoms in Molecules:A Quantum Theory[M].Oxford:Oxford University Press,1990.
[9]MATTA C F,BOYD R J.The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design[M].Weinheim:WILEY-VCH Verlag GmbH&Co KGaA,2007.
[10]POPELIER P L A.Atoms in Molecules:An Introduction[M].London:Prentice Hall,2000.
[11]GLENDENING E D,LANDIS C R,WEINHOLD F.NBO 6.0:natural bond orbital analysis program[J].Journal of Computational Chemistry,2013,34(16):1429-1437.
[12]WANG Q,ZHANG B,HUANG Z.Theoretical study on H2Y center dot center dot center dot Ag-X(X=F,Cl,Br,I;Y=O,S)complexes:structures,energies and bonding[J].Chemical Physics Letters,2014,6(14):145-149.
[13]SU P F,LI H.Energy decomposition analysis of covalent bonds and intermolecular interactions[J].J Chem Phys,2009,131(1):014102.
[14]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian09. Wallingford CT:Gaussian,Inc.,2009.
[15]BIEGLER-KÖNIG F,SCHÖNBOHM J.AIM2000.1.0 ed.Bielefeld,Germany:University of Applied Sciences,2000.
[16]LASINSKI M E,ROMERO N A,BROWN S T,et al.Recent performance improvements to the DFT and TDDFT in GAMESS[J].Journal of Computional Chemistry,2012,33(7):723-731.
[17]GUEST M F,BUSH I J,VAN DAM H J J,et al.The GAMESS-UK electronic structure package:algorithms,developments and applications[J].Molecular Physics,2005,103(6/7/8):719-747.
[18]WANG H K,HUANG Z G,SHEN T T,et al.Theoretical study on the hydrogen bonding interactions in 1∶1 supermolecular complexes of noradrenaline with water[J].Struct Chem,2012,23(4):1163-1172.
[19]WANG H K,HUANG Z G,SHEN T T,et al.Hydrogen-bonding interactions in adrenaline-water complexes:DFT and QTAIM studies of structures,properties,and topologies[J].J Mol Model,2012,18(7): 3113-3123.
[20]YU L,WANG Y H,HUANG Z G,et al.Structures,vibrational frequencies,topologies,and energies of hydrogen bonds in cysteine-formaldehyde complexes[J].Int J Quantum Chem,2012,112(5):1514-1525.
[21]NIU X Q,HUANG Z G,MA L L,et al.Density functional theory,natural bond orbital and quantum theory of atoms in molecule analyses on the hydrogen bonding interactions in tryptophan-water complexes[J]. J Chem Sci,2013,125(4):949-958.
[22]TIAN S X.Quantum chemistry studies of glycine-H2O2complexes[J].J Phys Chem B,2004,108(52):20388-20396.
[23]GALVEZ O,GOMEZ P C,PACIOS L F.Variation with the intermolecular distance of properties dependent on the electron density in cyclic dimerswithtwohydrogenbonds[J].J Chem Phys,2003,118(11):4878-4895.
[24]PARREIRA R L T,VALDES H,GALEMBECK S E.Computational studyofformamide-watercomplexesusingtheSAPTandAIMmethods[J]. Chem Phys,2006,331(1):96-110.
[25]KOCH U,POPELIER P L A.Characterization of C—H—O hydrogen bonds on the basis of the charge density[J].J Phys Chem,1995,99(24):9747-9754.
(责任编校 纪翠荣)
Theoretical study on the role of hydrogen bonds in acetaminophen-water complexes
YUAN Yuan,SUN Le,LI Yuying,WANG Xiaohong,HUANG Zhengguo
(a.College of Chemistry,b.Key Laboratory of Inorganic-Organic Hybrid Functional Materials Chemistry,Ministry of Education,c.Tianjin Key Laboratory of Structure and Performance for Functional Molecules,Tianjin Normal University,Tianjin 300387,China)
To learn the conformation of acetaminophen in water,six acetaminophen-water(AP-H2O)complexes formed by various types of hydrogen bonds(H-bonds)were characterized by geometries,energies,vibrational frequencies at the MP2/ 6-311++G(d,p)level.Natural bond orbital(NBO),quantum theory of atoms in molecules(QTAIM)analyses and the localized molecular orbital energy decomposition analysis(LMO-EDA)were performed to explore the nature of the hydrogen bonds in these complexes.The results showed that:(1)The intramolecular H-bond C8H8A…O2Aformed between the—CH and carbonyl oxygen atom of AP is retained in most of complexes;(2)The intermolecular H-bonds in AW1 and AW6 are stronger than the other intermolecular H-bonds;(3)The carbonyl oxygen atom of AP is the most likely site which can form H-bond with H2O molecule,and the second most likely site forming H-bond with H2O is the phenolic hydroxyl group of AP. Moreover,in the former case H-bonds are formed by the donation of proton from H2O to AP,while H2O acts as proton acceptor to form H-bond in the latter;(4)Both the hydrogen bonding interaction and structural deformation can determine the relative stabilities of AP-H2O complexes in some degrees,but the former had more important roles than the latter.
acetaminophen;complex;hydrogen bonding interaction;natural bond orbital(NBO);quantum theory of atoms in molecules(QTAIM)
O64
A
1671-1114(2016)04-0039-06
2016-03-12
天津市自然科学基金资助项目(12JCYBJC13400);天津市高等学校创新团队培养计划资助项目(TD12-5038).
原 媛(1991—),女,硕士研究生.
黄正国(1972—),男,副教授,主要从事计算化学方面的研究.
猜你喜欢
发明与创新(2023年4期)2023-01-10
中国医药导报(2021年35期)2022-01-20
椰城(2021年12期)2021-12-10
波谱学杂志(2021年3期)2021-09-07
少儿科学周刊·少年版(2021年22期)2021-01-17
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
华南师范大学学报(自然科学版)(2020年3期)2020-07-01
腐蚀与防护(2016年7期)2016-09-14
中学化学(2015年12期)2016-01-19
郑州大学学报(医学版)(2015年2期)2015-02-27
