
硫化氢直接分解制取氢气和硫黄研究进展
2017-04-07 10:27张婧张铁孙峰徐伟石宁
化工进展 2017年4期
张婧,张铁,孙峰,徐伟,石宁
(中国石油化工股份有限公司青岛安全工程研究院化学品安全控制国家重点实验室,山东 青岛 266071)
硫化氢直接分解制取氢气和硫黄研究进展
张婧,张铁,孙峰,徐伟,石宁
(中国石油化工股份有限公司青岛安全工程研究院化学品安全控制国家重点实验室,山东 青岛 266071)
硫化氢直接分解制取氢气和硫黄,不仅可以使石油、天然气、煤和矿产加工等生产过程中产生的硫化氢得到有效治理、解决环境污染问题,还能在回收硫黄的同时获得清洁能源——氢能。本文综述了热分解(直接热分解、催化热分解、超绝热分解)、电化学分解、光催化分解以及等离子体分解等硫化氢直接分解制取氢气和硫黄技术,对各种方法的基本原理、热力学依据进行了简要介绍,并详细阐述了各种技术的国内外研究现状,从研究方法、技术特点、反应性能、优缺点以及这些技术未来研究的可能突破点等方面展开深入分析。最后对硫化氢直接分解制氢技术的发展方向进行了展望,指出将膜技术、催化技术及等离子体技术相结合,不断发展和探索新技术将是硫化氢分解制氢技术的未来发展趋势。
制氢;热解;回收
硫化氢(H2S)是一种剧毒、恶臭的酸性气体,不仅会引起金属等材料的腐蚀,在化工生产中还容易导致催化剂中毒失活;另外,H2S还会危害人体健康,造成环境污染。因此,对石油、天然气、煤和矿产加工等工业领域中产生的大量H2S气体进行无害化处理技术,已经成为人们关注的热点。而传统克劳斯处理方法虽然可以将H2S部分氧化得到硫黄和水,然而却损失了大量氢资源。随着我国高硫原油加工量的增多,炼油加氢精制单元副产的含H2S酸性尾气量逐年增加,加氢精制所需的氢气量也随之增加;另外,氢气作为油品加氢裂化、低碳醇合成、合成氨等化工工艺过程的主要原料,其需求量也非常可观。因此,将H2S直接分解是一条理想的H2S资源化利用技术路线,既可以使其无害化,又可以生产氢气和单质硫,不仅可以实现氢资源在石油加工过程的循环利用,还可以减少传统烃类重整制氢带来的大量二氧化碳排放。
H2S直接分解制取氢气和硫黄一直是国内外科研工作者关注的研究领域,至今已报道了大量关于H2S分解的研究工作。H2S直接分解制取氢气和硫黄的方法主要包括:热分解法[1-3]、电化学分解法[4-6]、光催化分解法[7-10]和等离子体法[11-17]等。本文根据分解方法对于H2S分解制取氢气和硫黄的工艺路线进行了阐述,并对比分析了各种方法的特点,以期为H2S的综合回收利用提供一些思路。
1 热分解法
1.1 直接高温热分解法
直接高温热分解是指在无催化剂存在的条件下,通过高温直接将H2S热分解为氢气和硫黄。对于反应2H2S(g) —→2H2(g)+S2(g)而言,在标准状态下反应的ΔHӨ=171.6kJ,ΔSӨ=0.078kJ/K,ΔGӨ=148.3kJ>0,当无非体积功作用于反应体系时,该反应在常温常压下不能自发进行,而由热力学第二定律ΔGӨ=ΔHӨ–TΔSӨ可知,在高温条件下可使ΔGӨ<0,因此人们最初尝试使用高温热解法进行硫化氢的直接分解研究。SLIMANE等[18]在研究纯H2S高温热分解反应时发现,当温度低于850℃时,H2S几乎不发生分解反应,当温度分别为1000℃和1200℃时,H2S的转化率也分别只有20%和38%,当温度超过1375℃时,H2S的转化率才能达到50%以上(可根据ΔGӨ(T)=ΔHӨ(T)–TΔSӨ(T)=[ΔHӨ(298K) +及计算不同温度下分解反应所涉及的相关热力学数据,纯硫化氢分解反应随温度变化的曲线参见图1)。

图1 纯硫化氢分解反应随温度变化曲线[18]
由于2H2S(g) —→2H2(g)+S2(g) 反应受热力学限制,直接热分解难以达到较高的H2S转化率,因此研究者尝试通过引起化学平衡移动的方法来促进H2S的转化。例如,FARAJI等[19]通过降低分压的方法促进H2S的热分解反应。结果表明,在1200℃、1atm(1atm=101325Pa)、流速50mL/min和停留时间48s的条件下,纯H2S热分解转化率仅为35.6%,而当H2S分压降低至0.050atm时,1200℃条件下H2S转化率可增加至65.8%。
另外,也有研究者尝试通过移除产物引起化学平衡移动的方法来促进H2S的转化反应。FUKUDA等[20]使用耐热玻璃制成了一个封闭的循环系统,通过磁力泵实现反应气在反应池内的循环,利用冷阱去除热分解产生的硫黄,从而打破原有的化学平衡,提高H2S转化率。KAMEYAMA等[21-24]分别采用Vycor玻璃和氧化铝膜反应器进行了H2S分解反应研究。由于膜允许产物氢优先透过,使化学平衡发生移动,H2S转化率可提高两倍。Vycor玻璃膜孔径为4.5nm,膜进料侧的压力为405kPa、渗透侧压力为101kPa,稳定性实验表明该玻璃膜在600~800℃下可持续操作216h,并未发现膜结构破坏。但当反应温度高于800℃时膜结构会发生萎缩。而使用氧化铝膜反应器时,其操作温度可高于800℃,且渗透率是Vycor玻璃膜的30倍,但缺点是氧化铝膜的氢气分离因子很低。EDLUND等[25]也将H2渗透金属复合膜用于H2S的热分解反应,其方法是将具有Pt涂层的金属膜置于渗透侧,从而在反应温度为700℃、H2S分压为79.3kPa条件下能够有效地避免H2S对膜的化学腐蚀,实验结果表明在该膜反应器中H2S的转化率可达99.4%,接近完全转化。
采用直接高温热分解法,通过提高反应温度和降低H2S分压可以提高H2S转化率,但是该工艺需要供给大量热量、能耗高,并需要采用耐高温材料,所能处理的H2S浓度太低,不利于应用。此外,由于大量H2S需与H2分离,在系统中循环,增加了能耗。因此,此法在经济上受到严重的制约。采用膜技术虽然可以有效地分离产物从而打破化学平衡限制,提高H2S转化率,但热分解温度往往会超过膜的极限耐热温度,使膜材料结构遭到破坏,因此开发耐高温、分离效果好的膜材料是此技术未来发展的关键。
1.2 催化热分解法
催化热分解法是在热分解过程中加入催化剂进行热分解反应,加入催化剂虽然不能改变反应的热力学平衡,但可降低热分解反应的活化能,使H2S在较低的温度下便可发生分解反应,加快化学反应速率,提高H2收率。
硫化氢的分解反应属于氧化还原反应,因此目前研究中常用的催化剂为Fe、Al、V、Mo等过渡金属的氧化物或硫化物。张谊华等[26]使用不同方法制备了几种FeS催化剂,并研究了其对 H2S 分解制氢性能的影响。实验结果表明,以机械混合的超细粒子α-Fe2O3和γ-Al2O3为催化剂先驱物硫化制得的催化剂H2S分解反应性能最佳:反应温度为300℃时,其氢气收率可超过10%。
RESHETENKO等[27]在500~900℃温度范围内研究了γ-Al2O3、α-Fe2O3和V2O5催化剂对H2S多相热催化分解反应的影响。结果表明,H2S在γ-Al2O3和V2O5催化剂上的分解反应级数为2.0,而在α-Fe2O3催化剂上的分解反应级数是2.6,反应的有效活化能分别为72kJ/mol、94kJ/mol和103kJ/mol。研究还发现,在低温下H2S与γ-Al2O3相互作用先转化为HS–和S2–,温度升高后再进一步转化生成各种形态的单质硫。而H2S在α-Fe2O3和V2O5催化剂上的分解反应过程是将氧化物还原,同时形成Fe2+和V4+的硫化物。3种催化剂中α-Fe2O3催化剂的H2S分解效果最好,其在900℃时氢气收率可达31%左右。
GULDAL等[3]制备了3种钙钛矿结构催化剂,即LaSr0.5Mo0.5O3、LaSr0.5V0.5O3、LaMoO3,用于催化热分解H2S产生氢气和硫黄,实验发现在700~850℃温度范围内3种催化剂的催化活性从大到小的顺序为LaSr0.5Mo0.5O3>LaSr0.5V0.5O3>LaMoO3,而当温度提高至850~900℃时催化活性从大到小的顺序为LaSr0.5V0.5O3>LaSr0.5Mo0.5O3>LaMoO3,在950℃时使用LaSr0.5V0.5O3催化剂可获得最大H2S转化率为37.7%。
RICARDO等[28]设计了一种膜式催化反应器用于H2S分解制取氢气和硫黄,其方法是将MoS2催化剂沉积在管状陶瓷多孔膜元件上,该元件不仅能将H2S催化分解成氢气和硫黄,而且多孔陶瓷膜可以将产物氢选择性分离,从而引起化学平衡移动,促进分解反应的进行。在400~700℃、50.5~101kPa(0.5~1atm)条件下,处理含H2S为4%的混合气体,H2S的转化率可达56%,而在同样条件下仅用催化剂所获得的转化率只有40%。
催化剂可以降低反应的活化能,因而可以提高H2S 在低温下的热分解率,但催化剂的引入不能改变化学平衡,引入催化膜反应器可以在提高H2S转化率的同时将产物氢分离,进而提高反应转化率。因此而将催化剂与膜反应器或者其他促进分解反应平衡移动的装置相结合是今后研究的方向,然而该法的挑战在于更高效的催化剂制备和耐高温、低成本膜材料的研究开发。
另外,STARTSEV等[29-32]在溶剂层中使用Pt负载的SiO2/Al2O3/Sibunit及片状不锈钢作为催化剂进行了液固相低温催化分解H2S的反应研究。实验研究表明,当H2S/Ar混合物直接流过催化剂床层时,其转化率不超过5%,而当催化剂置于溶剂中时其转化率则显著提高:以NaCO3溶液作为溶剂时其硫化氢转化率可达到79.6%,当使用稀释的乙醇胺或肼作为溶剂时,硫化氢转化率则可达到98%以上。此方法可达到较高的硫化氢分解效率,但如何从溶剂中分离生成的硫黄将面临很大的技术挑战,溶剂处理困难将是其走向工业规模的限制因素。因此,寻找合适的溶剂来获得较高硫化氢分解效率将是解决这一问题的关键。
1.3 超绝热分解法
超绝热分解法是在无外加热源和催化剂的条件下,利用H2S在多孔介质中超绝热燃烧的方法实现H2S分解,其分解所需热量来自自身的部分氧化反应,无需额外的热源供给,利用该方法可有效解决H2S热分解过程能耗过高的问题。
BINGUE等[18,33]利用一种惰性多孔陶瓷介质研究了H2S-N2-O2混合气体(H2S含量为20%)中H2S的分解情况。实验结果表明,当量比(实际供氧量与理论完全燃烧1mol H2S需氧量)在0.1~5.5范围内H2S均可稳定燃烧,随着当量比的增加,H2收率呈现先增加后降低的趋势:当量比为2时,其燃烧温度接近1400℃,此时H2收率最大,接近20%。随着当量比的继续增加H2收率逐渐下降,当量比为5.5时不再产生氢气。
SLIMANE等[34]基于吉布斯自由能最小的原理,使用一种H2S部分氧化热力学模型,通过改变反应混合物进料组成的方法研究了H2S部分氧化条件下的超绝热分解情况。研究表明,当量比>6有利于H2的产生,当使用纯氧作为氧化剂时,其热力学最佳当量比>12时可在相对较低的热再生效率下获得较高的H2收率和较低的SO2收率。
凌忠钱等[35-36]采用计算流体力学数值模拟与化学动力学研究相结合的方法,使用标准湍流模型和一个17组分、57步复杂化学反应机理,对H2S在多孔Al2O3介质中的超绝热燃烧过程进行了研究。研究发现,富燃条件下H2S的燃烧温度比绝热燃烧温度高260℃,从而为H2S部分裂解提供了高温条件,将其裂解生成单质硫和H2。另外,该作者[37]还通过实验研究考察了H2S在反应器中停留时间以及反应温度等因素对裂解转化率的影响。实验发现,较长的停留时间、较高的反应温度以及较低的 H2S初始浓度,有助于获得较高的H2S转化率。
超绝热分解法使用多孔介质为H2S分解提供富燃条件,并通过H2S的部分氧化为其分解提供能量,使H2S的燃烧温度超过绝热燃烧温度,避免了外加热源的引入,极大地降低了H2S热分解能耗。此外,超绝热分解过程还具有NOx、CO、SO2排放量小的优点。然而,超绝热分解法要求反应器填充的多孔介质能耐1400~1600℃的高温,因此,开发蓄热能力强、透气性能好、耐热耐酸、成本低廉的多孔材料是该技术的关键。
2 电化学分解法
由前述热力学分析可知:当无非体积功作用于反应体系,ΔGӨ<0时反应在常温常压下不能自发进行;但当外界对反应体系做功,使ΔGӨ–W<0,则反应仍能发生,电化学分解法则是在电解槽中利用电能对体系做功,将H2S直接分解得到氢气和硫黄。电化分解法分为直接电解和间接电解两种。由于H2S在水中的溶解度较低,因此通常采用电解H2S碱性溶液的方法来制取氢气和硫黄。ANANI等[5]在80℃下使用等摩尔浓度NaOH、NaHS混合碱液进行了H2S电解研究,其产物收率可达到95%以上。SHIH等[38]以甲苯、Na2S和NaOH的混合液作为电解液,在连续搅拌槽式电化学反应器中进行了H2S电解制取氢和硫黄的研究,在甲苯流量2~13mL/min、Na2S-NaOH(Na2S∶NaOH摩尔比为1∶ 50)流量10~66mL/min、温度20~60℃、外加电压1.8~4.8V条件下,获得的硫收率约为40%~80%。然而,直接法电解H2S,生成的硫黄会沉积在阳极表面,导致电极钝化,因此后来的研究主要集中在间接法电解H2S上。
间接电解法通常是使用一种氧化剂作为中间循环剂,以氧化还原反应和电解反应共同构成双反应工艺。KALINA等[6,39]在高电流密度和电流效率条件下,使用碘酸钾作为中间循环剂,通过电化学方法在阳极将氢碘酸溶液中I–氧化成I3–,同时阴极生成H2;而强碱性溶液中I–与H2O反应生成IO3–,IO3–与H2S发生反应生成硫黄,从而完成氧化还原反应的电化学循环;然而,此法产生氢气的同时会生成可溶性I3–,这种I3–可以与H2S发生反应导致生成的硫黄纯度下降。
MIZUTA等[4]采用Fe3+/Fe2+作为中间循环剂进行了间接电解H2S的研究。该反应过程中H2S首先与FeCl3溶液反应生成硫黄、FeCl2和HCl,然后将去除硫黄后的FeCl2+HCl溶液进行电解,Fe2+在阳极上被氧化成Fe3+,H+在阴极上被还原生成氢气;实验结果表明:在温度70℃、电解电压0.7V条件下,H2S的转化率可达到接近100%;此外,MIZUTA等还建立了包含H2S吸附单元、硫黄分离单元和电解单元的小试装置,其氢气日产量为2.1m3/d,硫黄日产量为3kg/d。
罗文利等[40]也使用含Fe3+的强酸性反应液进行了H2S的吸附、电解反应研究,在实验条件下H2S的吸收率可达85%,电解制氢和氧化液再生均可在1.2V的低电压下进行,其电流效率均接近100%。
李发永等[41]采用制氢规模为100~120L/h的扩大实验装置对体积分数为85%~95%的H2S进行了间接电解研究,结果发现当吸收温度为60℃、吸收液中Fe3+浓度大于2.5mol/L、液气体积比大于1.5时,H2S的吸收率可超过99%,制得的硫黄纯度高于99.8%,而此时制氢电耗约为 2.9kW·h/m3。
袁长忠等[42]对H2S间接电解制氢中试的阴极传质过程和电化学过程进行了研究,建立了阴极析氢过程宏观动力学模型,该模型在测量范围内与实验数据吻合较好,并发现H2S间接电解制氢阴极反应的主要影响因素为H+浓度、槽压和电解液温度,其中槽压的影响最大。
间接电解法可处理高浓度的H2S气体,并且硫黄回收率高、制氢单位电耗较低。但该方法还存在传质效率低、电极材料的使用寿命短等缺点。因此研发长寿命、高性能、低价格的电极材料和电解装置,开发高效中间循环氧化剂是该工艺应用的关键。
3 光催化分解法
光催化法分解H2S通常是采用具有非连续能级的半导体材料作为催化剂,当具有大于半导体材料禁带能级能量的光子照射半导体材料时,其价带电子就会被激发,并跃迁至导带,从而形成电子-空穴对,电子-空穴对与H2S相互作用发生光催化氧化还原反应,进而分解产生氢气和硫黄。这种方法也是利用光能对反应体系做非体积功,光能转化为化学能,使得ΔGӨ–W<0,进而促使反应发生,其反应原理如式(1)~式(3)。

目前,研究较多的光催化剂主要有金属硫化物光催化剂、复合光催化剂以及负载型光催化剂等几类。一些光催化剂分解H2S的制氢性能对比如表1所示。

表1 光催化剂分解H2S制氢性能对比
光催化分解H2S所使用的金属硫化物催化剂大多是以ZnS、CdS等半导体为主要活性组分。其中ZnS的禁带约为3.6eV,只能被小于360nm的紫外光激发;CdS具有较低的禁带宽度,对紫外和可见光均有响应。SERPONE等[43-44]考察了负载RuO2、Pt以及TiO2的CdS半导体光催化剂分解H2S制氢的性能。结果表明,负载少量RuO2可显著提高CdS的光催化活性。在浓度为0.1mol/L Na2S-1.0mol/L NaOH混合溶液中,当反应温度为40℃,使用λ> 400nm的可见光照射,以1.0% RuO2/CdS作为催化剂时(质量分数),其产氢速率可达1.86mL/h,然而当以0.5%RuO2-TiO2/CdS作为催化剂时,其产氢速率可进一步提高至2.23mL/h,其催化活性约是体相CdS的5.4倍。
KHAN等[45]将Pt负载于CdS、CdS/Ag2S和CdxZnxS/Ag2S,制备出H2S分解光催化剂,实验结果表明CdxZnxS/Ag2S/Pt催化剂具有最优的H2S分解制氢性能,当Pt、Ag2S和ZnS负载量为2%(质量分数)、1%和20%(摩尔分数)时具有最高的催化活性。
DE等[46]将Ag2S和Pt先后负载于CdS上,再将其与ZnS混合制得Pt/CdS/Ag2S-ZnS光催化剂,并以Na2S-Na2SO3混合溶液为反应液,进行了光催化分解H2S制氢的研究。在16h的太阳光照射下,获得的平均产氢约为1.53mL/h。此外,为了促进电子与空穴的分离,研究者在反应中加入0.2~0.6V的偏电压,偏电压的加入可将产氢速率提高10%~20%。
马贵军等[47]使用TiO2、CdS、ZnS、ZnO以及ZnIn2S4等半导体光催化剂进行了气固相直接分解H2S制氢的研究。实验结果表明,在ZnS上负载贵金属Ir可明显提高产氢速率,而在制备ZnS 的过程中加入少量Cu2+则可大幅度提高其光催化分解H2S的活性,当加入Cu2+摩尔分数为0.5%时,在λ>420nm的可见光照射下产氢速率可达17μmol/h。
过渡金属氧化物由于其良好的光催化活性及稳定性被广泛用于光催化领域,但其禁带宽度较高,仅能吸收紫外光,对太阳光利用率低。为此,研究者们将金属氧化物和硫化物相结合制备出复合光催化剂,使其具有更好的可见光响应性能和更优越的光催化活性。SO等[48]将纳米CdS与TiO2溶胶按照不同摩尔比机械混合、通过水热法制成CdS-TiO2复合微粒薄膜,再使用TiCl4水溶液对薄膜进行表面处理,成功制备出 CdS-TiO2复合光催化剂。实验结果表明:当CdS与TiO2摩尔比为1∶4时具有最高的产氢速率,约为1.8 mL/(cm2·h)。
JANG等[49]使用纳米TiO2颗粒修饰高结晶度的CdS,制备出比表面积约为97m2/g的CdS/TiO2光催化剂。该催化剂的禁带宽度约为2.25eV,在λ≥420nm可见光照射下其产氢速率可达422.4μmol/h。
RUBAN等[50]合成了一种核-壳结构的(CdS-ZnS)TiO2纳米颗粒,并将其用于光催化分解H2S制取氢气的反应。实验发现,与CdS、ZnS、CdS-ZnS和TiO2纳米颗粒相比,具有核-壳结构的(CdS-ZnS)TiO2纳米颗粒具有最高的产氢速率(29mL/h)。此外,还研究了S2–浓度、SO32–浓度、pH、催化剂浓度等对核-壳结构的(CdS-ZnS)TiO2纳米颗粒催化性能的影响,实验发现在0.05mol/L S2–浓度、0.2mol/L SO32–浓度、pH为11.3和500mg/L光催化剂条件下能获得最大速率常数0.0038min–1,在优化的实验条件下H2S的转化率可达30%。
此外,多元过渡金属形成的复合光催化剂也具有良好的H2S分解性能。SUBRAMANIAN等[51]使用传统固相反应法制备出了ZnFe2Ta2O9光催化剂,并在λ≥420nm可见光条件下进行了H2S分解反应研究。在0.5mol/L KOH溶液中,当H2S通入速度为2.5mL/min时,单次焙烧的ZnFe2Ta2O9光催化剂产氢速率能达到1657μmol/h,二次焙烧的ZnFe2Ta2O9光催化剂产氢速率更高达2320μmol/h,两种催化剂的量子效率分别为8.1%和11.4%。
负载型光催化剂是将具有光催化活性的半导体担载于Al2O3、SiO2、高分子聚合物或分子筛等载体,从而形成的催化剂。由于其具有较高的比表面积,因此可以进行更有效的电子传递和离子交换。KANADE等[52]通过CdS与聚苯硫醚(PPS)之间的聚合物无机固态反应,合成出6~28nm尺寸的CdS晶体,并将其嵌入到聚苯硫醚(PPS)骨架中,制备出纳米CdS-PPS光催化剂。将0.5g此催化剂置于250mL 0.5mol/L的KOH溶液中,在λ≥500nm的可见光照射下,通入2.5mL/min H2S进行光催化反应,催化剂的光量子效率可达19.7%,得到的最高制氢速率为3926μmol/h,约为Pt-CdS催化剂的2.6倍。
BAI等[9]采用浸渍法将纳米CdS负载于SiO2/Al2O3比为3.5的HY沸石分子筛,制备出CdS/HY光催化剂。研究表明,CdS团簇不仅存在于沸石分子筛孔内,还聚集在沸石分子筛笼的外表面,HY沸石分子筛能够有效的保护CdS纳米颗粒免于光腐蚀。与体相CdS相比,CdS/HY光催化剂具有更高的光催化活性,其制氢速率为24mmol/g h,比体相CdS的制氢速率高5.38倍。
WANG等[53]使用一步法合成了高活性Ti-Cr-MCM-48(Si/Ti=3.4,Si/Cr=50)介孔材料,并将其用于硫化氢光催化分解反应。H2S-N2-O2混合物以50mL/min(H2S体积分数为0.0035%)通入反应器,使用可见光照射5min后H2S转化率便可达到73%,照射60min后H2S转化率则增加到92%左右,然而继续增加照射时间H2S转化率则显著降低。研究发现,Cr6+为光催化反应主要活性物种,而硫酸盐在催化剂表面的累积以及Cr6+的减少是导致催化剂失活的主要原因。
另外,ZONG等[10]通过使用Fe2+/Fe3+和I–/I3–氧化还原电对构成的光化学-化学循环进行了光电催化H2S分解制氢的反应研究。在使用功能化的Si材料作光电极,在模拟太阳光的照射下获得较高的H2选择性和稳定性,其产氢速率可达90~120μmol/h。该方法首次实现光电化学过程一步完全分解H2S,而不需要对于硫基水溶液进行后处理。
光催化和光电催化法利用廉价丰富的太阳能作为能量来源,反应条件温和、能耗低,是一种较为经济的方法。但该方法存在光子利用效率低、可见光响应催化剂制备成本高、回收困难,催化剂容易光腐蚀、中毒等缺点。目前光催化分解H2S的研究主要集中在光催化剂的设计与研究开发以及反应工艺过程的设计和优化等方面。开发具有可见光响应、较高光子利用效率、较低光生空穴与电子复合几率的廉价光催化剂,以及设计合理的工艺路线是光催化分解H2S研究的关键。
4 等离子体法
等离子体被称为物质存在的第四种状态,其中包含大量的基态分子、原子、激发态分子、原子、离子、自由基、电子和光子等,其宏观呈电中性。低温等离子体由于具有较高的电子温度和较低的气体温度,可以在常温状态下有效的活化反应物分子,而被广泛用于化学反应领域[54]。等离子体法是通过电能引发反应物气体放电,从而对反应体系做非体积功,使得ΔGӨ–W<0,进而促使反应发生。目前,多种放电方法被用于H2S分解制取氢气和硫黄,主要有辉光放电、电晕放电、介质阻挡放电、滑动电弧放电、射频等离子和微波等离子体等。由于不同的放电形式具有不同的平均电子能量、放电特性、击穿电压、电压-电流特征等不同的物理和电学性质,因此对不同放电形式分解H2S进行分别介绍。一些等离子体法分解H2S的转化效率对比如表2所示。
4.1 辉光放电
辉光放电是气体一种稳定的自持放电形式,其击穿机制是从阴极发射电子,从而在放电空间引起电子雪崩,由此产生的正离子再轰击阴极致使其发射出更多的电子,是由电子雪崩的不断发展引发的放电。
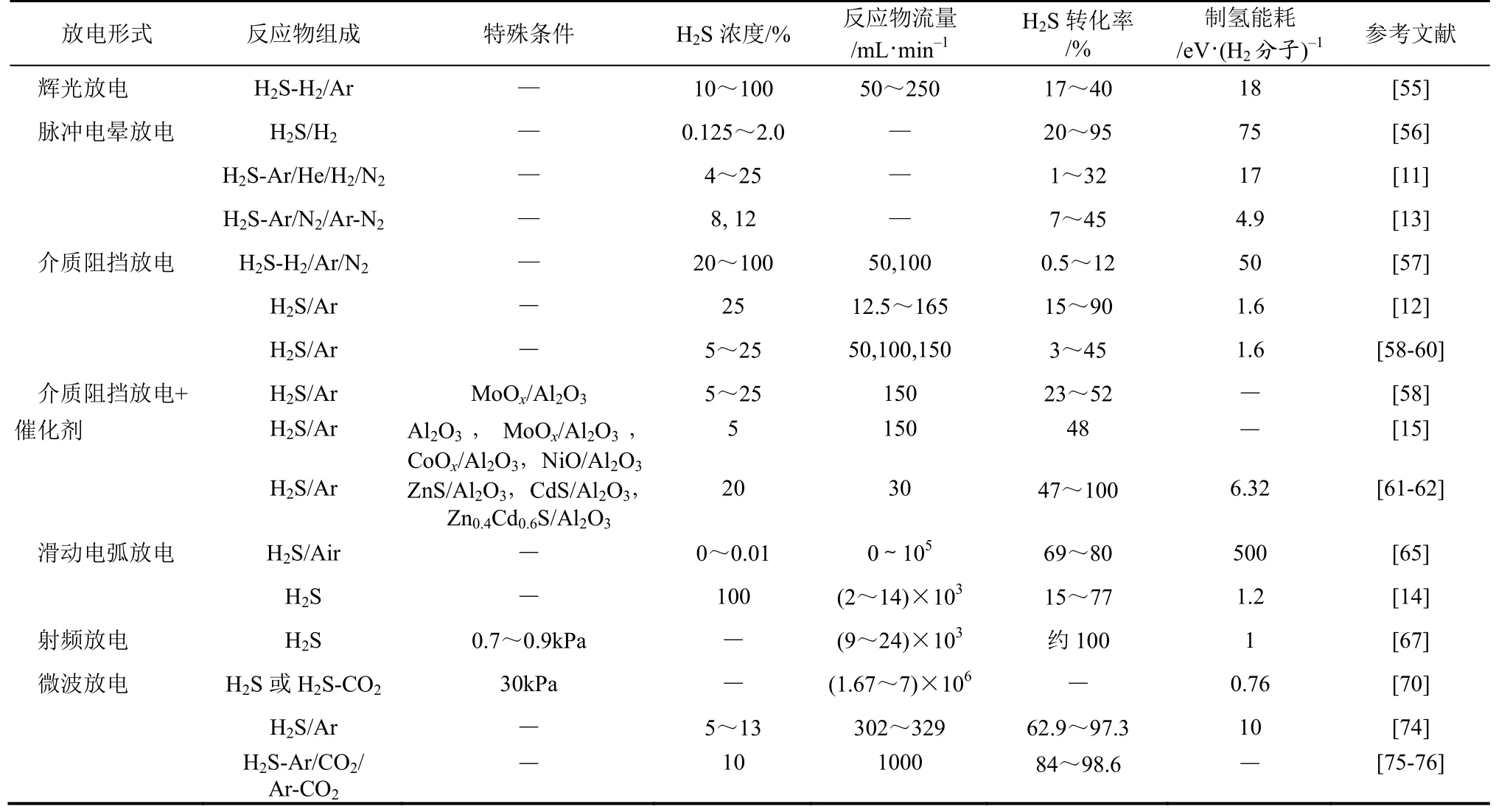
表2 等离子体法分解H2S
TRAUS等[55]使用旋转高压辉光放电分解H2S得到氢气和硫黄,将同轴电极置于磁场中,并通过电极加热的方式有效地去除生成的硫黄,从而实现持续稳定放电。考察了放电电流和磁感应强度对转动频率的影响,并考察了注入功率、转动频率、进料流量、H2S浓度对H2S转化率和能量效率的影响。在H2S初始浓度10%~100%(H2或Ar作为添加气)、进料流量为100mL/min、放电间隙3.5mm、电极温度170℃、放电功率45~75W条件下得到H2S转化率为17%~40%,最低制氢能耗为2.1mol/(kW·h)(约18eV/H2分子)。
4.2 电晕放电
电晕放电是气体在不均匀电场中发生的局部自持放电,当极高的电压加到电极两端,在曲率半径小的电极附近就会形成较强的局部场强,从而使电极附近气体局部击穿,形成电晕放电。电晕放电根据电源的性质不同可分为直流电晕和脉冲电晕两类,其中脉冲电晕为目前等离子体分解H2S研究广泛采用的一种放电形式。
HELFRITCH等[56]使用线管式脉冲电晕放电反应器研究了H2S的分解反应,考察了反应器结构、H2S浓度以及注入功率等对反应的影响,并建立数学模型对实验结果进行了预测。研究发现减小放电反应器直径有利于提高反应能效,此外,与处理静态气体相比,处理动态气体时可获得更高的能效。
ZHAO等[11]在Ar、He、N2、H2等平衡气存在下使用脉冲电晕放电进行了H2S分解制取氢气和硫黄的研究。在不同H2S浓度和压力下测量了H2S与Ar、He、N2、H2共放电条件下的击穿电压、H2S转化率和分解能效,研究结果表明,加入平衡气可以有效地降低H2S击穿电压,且击穿电压正比于H2S在混合气中的分压。而H2S的转化率和分解能效与平衡气的种类和H2S浓度相关。与N2、H2等双原子气体作为平衡气相比,当Ar、He等单原子气体作为平衡气时,H2S的转化效率更高。在Ar作为平衡气、H2S浓度为16%时,得到最低制氢能耗约为17eV/H2分子。
JOHN等[13]采用脉冲电晕放电进行了H2S分解制取氢气和硫黄的研究,反应器采用线管式结构,在固定功率100W条件下考察了脉冲形成电容、放电电压以及脉冲频率对H2S转化率和分解能效的影响。结果表明,在功率一定的条件下,低脉冲形成电容、低放电电压以及高脉冲频率有利于获得高H2S分解能效;另外,与Ar和N2作为平衡气相比,以Ar-N2混合气作为平衡气时可以得到更高的H2S转化率,在Ar/N2/H2S体积分数为46%/46%/8%、放电功率60W、脉冲形成电容720pF时,获得的H2S最低分解能耗为4.9eV/H2S分子,但此时H2S转化率仅为30%左右。
4.3 介质阻挡放电
介质阻挡放电是有绝缘介质插入放电空间的一种气体放电状态。介质阻挡放电通常呈微通道放电结构,即通过放电间隙的电流由大量快脉冲电流细丝组成,电流细丝在时间和空间上均为随机分布。由于介质的存在,限制了放电电流的增长,从而避免了气体完全击穿形成火花或电弧,这有利于大体积、稳定等离子体的产生。由于介质阻挡放电具有均匀、稳定的特性,早在20世纪90年代初,科学工作者就开始使用其进行H2S分解制取氢气和硫黄的研究。TRAUS等[57]使用改进的臭氧发生器考察了H2S在130~560℃范围内的放电特性,并研究了反应温度、H2S进料浓度、注入功率以及添加H2、Ar、N2等对H2S转化率和能量效率的影响,得到的H2S转化率为0.5%~12%,最低产氢能耗约为0.75mol/(kW·h)(50eV/H2分子),实验发现添加Ar能够促进H2S的分解,得到更高的H2S转化率和能量效率。
SUBRAHMANYA等[12]采用烧结的金属纤维作为内电极,通过介质阻挡放电的方式分解H2S制取氢气,金属纤维同时兼做反应的催化剂,考察了注入能量密度、电极间隙、放电频率和气体停留时间对H2S分解的影响,当以H2S/Ar混合气(H2S体积分数为25%)作为反应气,在气体停留时间为6s时H2S分解制氢最低能耗可达到160kJ/mol H2(约1.6eV/H2分子)。
REDDY等[58-60]以Ar为载气使用介质阻挡放电进行了H2S分解制氢的研究。对放电间隙、气体在放电区的停留时间、放电频率、H2S初始浓度和温度等条件进行了优化考察,并使用不同接地电极材质(银涂层、铜线、铝箔)进行了H2S分解实验,实验发现接地电极材质的改变仅仅增加了能量注入,并没有显著改善转化率,而其他反应条件对H2S的转化率及制氢性能都有较大影响,在最优实验条件下得到的最低制氢能耗为160kJ/mol(约1.6eV/H2分子)。
通过反应器结构和实验条件的优化,介质阻挡放电分解H2S的能耗大大降低,然而单纯介质阻挡放电仍难获得较高的H2S转化率,因此科研工作者们开始尝试将催化剂填充至放电区,通过等离子体和催化剂的协同作用进一步提高H2S转化率、降低制氢能耗。REDDY等[15]将Al2O3、MoOx/Al2O3、CoOx/Al2O3和NiO/Al2O3催化剂填充在放电区,考察了金属氧化物活性组分、催化剂填充长度及H2S初始浓度对H2S转化率的影响。反应结果表明MoOx/Al2O3和CoOx/Al2O3催化剂具有较好效果;其中当填充MoOx/Al2O3催化剂,注入比能SIE为0.92kJ/L、催化剂填充长度为床层10%时,得到的H2S最高转化率约为48%。
ZHAO等[61]将Al2O3负载的ZnS和CdS(ZnS/ Al2O3和CdS/Al2O3)颗粒填充于等离子体区,进行介质阻挡放电和半导体催化剂协同分解H2S的实验研究。当使用空管放电时,H2S的最高转化率仅为67%左右,此时H2S分解能耗约为18.20eV;而在等离子体区填充ZnS/Al2O3和CdS/Al2O3颗粒时,H2S完全分解的能耗分别为9.49eV和6.92eV。与单纯介质阻挡放电转化H2S相比,等离子体区填充ZnS/Al2O3和CdS/Al2O3催化剂能明显提高H2S的转化率,并能显著降低H2S的分解能耗。
ZHAO等[62]在另一篇文献中考察了比能量密度(SIE)、反应物流速、金属硫化物负载量以及加入H2对等离子体-催化协同分解H2S的影响。研究表明,Al2O3负载的ZnS和CdS(ZnS/Al2O3和CdS/Al2O3)与介质阻挡放电具有协同分解H2S的作用;此外,与ZnS/Al2O3和CdS/Al2O3相比Zn0.4Cd0.6S/ Al2O3催化剂具有更高的H2S分解活性,达到H2S完全分解所需的能耗更低(6.32 eV)。
ZHAO等[16]还报道了一种介质阻挡放电硫化制备高分散负载型硫化物的方法,并将介质阻挡放电硫化制备的ZnS/Al2O3、CdS/Al2O3催化剂与传统热硫化方法制备的ZnS/Al2O3、CdS/Al2O3催化剂进行对比。结果表明:等离子体硫化制备的ZnS/Al2O3和CdS/Al2O3催化剂颗粒尺寸更小、更均匀、分散度更好,达到H2S完全分解所需的能耗也更低。
4.4 滑动电弧放电
辉光放电、电晕放电、介质阻挡放电等低温等离子体能够产生较多的化学活性,但其功率和温度都较低,对于H2S转化这样高耗能过程,最好的办法是使用高功率和较高气体温度的等离子体[63],而滑动电弧放电具有高达2000~4000K的气体温度,而被认为是“温暖”低温等离子体[64],其具有较高的功率和气体温度,有利于化学转化,同时具有较低的能量损失。
DALAINE等[63,65]使用滑动弧光放电对H2S分解反应进行了研究,其方法是将H2S用空气稀释至浓度为0~100μL/L,在气体总流速为0~100L/min条件下测量了放电电压、电流、温度、速度和放电长度等特性参数,并考察了气体流动速率、反应腔体尺寸和频率对H2S分解反应的影响。实验结果表明低气体流速、小盘间距及低频率有利于获得较高的H2S转化率,在优化的放电条件下得到的H2S转化率可达75%~80%,但H2S分解能耗较高,约500eV/H2S分子。
NUNNALLY等[14,66]利用滑动电弧旋风等离子体放电反应器进行了H2S分解反应的研究,独特的反应器结构使得反应器壁处于低温状态,而反应器轴心位置具有较高的温度,该反应器延长了H2S在放电区的停留时间,并使其在等离子体中心区域内进行强制对流传热传质;另外,生成的硫原子簇在旋转气流所产生的离心作用下可迅速脱离反应区,从而达到猝灭逆反应、提高了H2S转化率的效果。考察了反应压力、气体流速对H2S转化率和分解能耗的影响,结果表明降低压力和气体流速有利于提高氢气的收率,但低压条件下H2S的分解能耗较高。在气体压力为1atm、H2S流速为14L/min、注入比能为0.31eV时,H2S的最低分解能耗约为1.2eV/H2S分子,但此时H2收率仅为25%左右。
4.5 射频等离子体
射频等离子体是利用高频高电压使电极周围的空气电离而产生的低温等离子体。射频等离子可以产生线形放电,也可以产生喷射形放电,现在已经被应用于材料的表面处理和有毒废物清除和裂解中,但将射频等离子体用于H2S分解研究的报道却较少。
KRASHENINNIKOV等[67]采用射频等离子体放电进行了H2S分解制氢的研究,研究中采用了多级射频等离子体反应器,达到的最高H2S转化率接近100%,但所需的能耗较高。在压力为700~900Pa、气体流量9~24L/min、注入功率0.6~2kW条件下,可获得较低的制氢能耗,其最低制氢能耗约为1eV/H2分子。极低的操作压力限制了其实际应用,并且难以实现高气速转化。
4.6 微波等离子体
微波等离子体是将微波能量转化为气体分子的内能,使之激发、电离从而产生等离子体的一种气体放电形式。采用微波等离子体进行放电时,电源发生的微波通过传输线传输到贮能元件,再以某种方式与放电管耦合,藉磁场能将能量赋予当做负载的放电气体,无需在放电空间设置电极而功率却可以局部集中,因此能获得高密度等离子体,在功率为千瓦级时其电子密度可接近等离子体频率所确定的临界密度,能够比一般放电提供更高的电离度和解离度。
前苏联ASISOV[68]和BAGAUTDINOV[69-72]等采用微波等离子体对H2S及H2S/CO2混合物(两者物质的量之比约为0.5~0.6)的分解反应进行了研究,并在小试实验的基础上建立了微波等离子体中试装置,在注入功率1MW、气体流量每小时数千立方米条件下,获得H2S分解能耗可低至0.76eV/ H2S分子左右。随后,受其鼓舞,亚伯达氢研究规划署、加拿大原子能署和Shell有限公司联合建立了类似装置,但未能达到其相对较低的能耗,得到的最低制氢能耗在4.5eV左右[73]。
国内中国科学院感光化学研究所和武汉工程大学的科研人员也使用微波等离子体进行了相关研究。董永治等[74]以Ar为反应载气,使用矩形共振腔微波反应器在常压下产生等离子体,考察了狭缝宽度、H2S气体流量对H2S分解反应的影响,在狭缝宽度为15mm、Ar流量287mL/min、H2S流量15mL/min时达到最低制氢能耗为0.48mol/(kW·h)(约10eV),此时H2S的转化率为92.0%,氢气的产生速率为13.8mL/min。
汪建华等[75]在Ar、CO2及Ar-CO2混合气体3种载气条件下采用常压微波等离子体对H2S进行了分解实验研究,考察了微波功率对H2S分解效率的影响。结果表明:在一定功率范围内(400~1100W)增加微波功率有利于提高H2S的分解效率,当微波功率继续增加时,不同的载气条件的分解效率变化趋势不同。在相同微波功率条件下,载气为Ar-CO2、Ar-CO2-H2S混合气体流量比为8∶1∶1、总流量为1000mL/min、微波功率为1300W时,H2S转化率最高可达98.64%。此外,他们[76]还在Ar作载气时使用大气微波等离子体射流装置对H2S气体进行了分解研究,考察了温度、微波功率、载气流量及气体总流量对H2S分解效率的影响。实验结果表明,低温、低气体总流量有利于H2S的分解,而微波功率和载气流量则存在一个最佳值。当H2S/Ar流量比为10∶90、气体总流量为1000mL/min、微波功率为1000W时,H2S的最高分解率可达91.32%。
在等离子体放电条件下,通过施加电能对反应体系做功,使得H2S可以在较低的温度下被激发、解离以及电离,产生各种激发态的原子、分子,自由基及高能电子等活性物种,通过活性物种间的相互碰撞引发化学键的断裂和重组,从而获得较高的转化率;目前,关于等离子体分解H2S有许多相关研究,但大多处于实验室研究阶段,主要集中在等离子体反应器的设计及放电参数的优化等,对反应机理的认识较少;此外,等离子体法要实现工业化还需解决能量效率高的大功率电源的研制、耐压耐腐蚀装置的开发等工程问题。另一方面,将催化剂引入低温等离子体反应体系,可显著提高H2S转化率,降低制氢能耗,但等离子体与催化剂协同转化H2S尚处于探索阶段、反应机理尚不清晰,两者的协同作用机制尚待深入研究。
5 结语
随着我国经济的飞速发展,能源、化工等领域产生的含H2S废气逐年增加,因此无论是从含H2S尾气治理避免环境污染还是以H2S分解制氢和硫以获得清洁能源出发,H2S直接分解制取氢气和硫黄技术均具有非常大的研究意义。目前发展的各种技术涉及多个学科,并且处于不同的发展阶段,各种工艺各有其优缺点,因此要找到一种反应条件温和、转化效率高、能耗低的工艺方法还需不断的研究探索;将膜技术、催化技术及等离子体技术相结合,不断发展和探索新技术,并将其与现有工艺相结合,将是H2S分解制氢技术未来的发展趋势。
[1]AL-SHAMMA L,NAMAN S. The production and separation of hydrogen and sulfur from thermal decomposition of hydrogen sulfide over vanadium oxide/sulfide catalysts [J]. Int. J. Hydrogen Energy,1990,15(1):1-5.
[2]AKAMATSU K,NAKANE M,SUGAWARA T,et al. Development of a membrane reactor for decomposing hydrogen sulfide into hydrogen using a high-performance amorphous silica membrane [J]. Journal of Membrane Science,2008,325(1):16-19.
[3]GULDAL N,FIGEN H,BAYKARA S. New catalysts for hydrogen production from H2S:preliminary results[J]. Int. J. Hydrogen Energy,2015,40(24):7452-7458.
[4]MIZUTA S,KONDO W,FUJII K,et al. Hydrogen production from hydrogen sulfide by the iron-chlorine hybrid process [J]. Industrial & Engineering Chemistry Research,1991,30(7):1601-1608.
[5]ANANI A,MAO Z,WHITE R E,et al. Electrochemical production of hydrogen and sulfur by low-temperature decomposition of hydrogen sulfide in an aqueous alkaline solution[J]. Journal of the Electrochemical Society,1990,137(9):2703-2709.
[6]KALINA D,MAAS JR E T. Indirect hydrogen sulfide conversion——Ⅰ. An acidic electrochemical process [J]. Int. J. Hydrogen Energy,1985,10(3):157-162.
[7]KALE B,BAEG J O,YOO J S,et al. Synthesis of a novel photocatalyst,ZnBiVO4,for the photodecomposition of H2S[J]. Canadian Journal of Chemistry,2005,83(6/7):527-532.
[8]YAN H,YANG J,MA G,et al. Visible-light-driven hydrogen production with extremely high quantum efficiency on Pt-PdS/CdS photocatalyst [J]. Journal of Catalysis,2009,266(2):165-168.
[9]BAI X F,CAO Y,WU W. Photocatalytic decomposition of H2S to produce H2over CdS nanoparticles formed in HY-zeolite pore [J]. Renewable Energy,2011,36(10):2589-2592.
[10]ZONG X,HAN J,SEGER B,et al. An integrated photoelectrochemical–chemical loop for solar-driven overall splitting of hydrogen sulfide[J]. Angewandte Chemie International Edition,2014,53(17):4399-4403.
[11]ZHAO G B,JOHN S,ZHANG J J,et al. Production of hydrogen and sulfur from hydrogen sulfide in a nonthermal-plasma pulsed corona discharge reactor [J]. Chemical Engineering Science,2007,62(8):2216-2227.
[12]SUBRAHMANYA M C,RENKEN A,KIWI MINSKER L. Non-thermal plasma catalytic reactor for hydrogen production by direct decomposition of H2S [J]. Journal of Optoelectronics and Advanced Materials,2008,10(8):1991-1993.
[13]JOHN S,HAMANN J C,MUKNAHALLIPATNA S S,et al. Energy efficiency of hydrogen sulfide decomposition in a pulsed corona discharge reactor [J]. Chemical Engineering Science,2009,64(23):4826-4834.
[14]NUNNALLY T,GUTSOL K,RABINOVICH A,et al. Dissociation of H2S in non-equilibrium gliding arc “tornado” discharge[J]. Int. J. Hydrogen Energy,2009,34(18):7618-7625.
[15]LINGA REDDY E,KARUPPIAH J,BIJU V,et al. Catalytic packed bed non-thermal plasma reactor for the extraction of hydrogen from hydrogen sulfide [J]. International Journal of Energy Research,2013,37(11):1280-1286.
[16]ZHAO L,WANG Y,SUN Z,et al. Synthesis of highly dispersed metal sulfide catalystsvialow temperature sulfidation in dielectric barrier discharge plasma [J]. Green Chemistry,2014,16(5):2619-2626.
[17]REDDY E L,BIJU V,SUBRAHMANYAM C. Hydrogen production from hydrogen sulfide in a packed-bed DBD reactor[J]. Int. J. Hydrogen Energy,2012,37(10):8217-8222.
[18]SLIMANE R B,LAU F S,DIHU R J,et al. Production of hydrogen by superadiabatic decomposition of hydrogen sulfide[C]//proceedings of the Proc 14th World Hydrogen Energy Conference,US:NREL,2002:1-15.
[19]FARAJI F,SAFARIK I,STRAUSZ O P,et al. The direct conversion of hydrogen sulfide to hydrogen and sulfur[J]. Int. J. Hydrogen Energy,1998,23(6):451-456.
[20]FUKUDA K,DOKIYA M,KAMEYAMA T,et al. Catalytic decomposition of hydrogen sulfide [J]. Industrial & Engineering Chemistry Fundamentals,1978,17(4):243-248.
[21]KAMEYAMA T,DOKIYA M,FUKUDA K,et al. Differential permeation of hydrogen sulfide through a microporous Vycor-type glass membrane in the separation system of hydrogen and hydrogen sulfide [J]. Separation Science and Technology,1979,14(10):953-957.
[22]KAMEYAMA T,FUKUDA K,FUJISHIGE M,et al. Production of hydrogen from hydrogen sulfide by means of selective diffusion membranes [J]. Hydrogen Energy Progress,1981,1:569-579.
[23]KAMEYAMA T,DOKIYA M,FUJISHIGE M,et al. Possibility for effective production of hydrogen from hydrogen sulfide by means of a porous Vycor glass membrane[J]. Industrial & Engineering Chemistry Fundamentals,1981,20(1):97-99.
[24]KAMEYAMA T,DOKIYA M,FUJISHIGE M,et al. Production of hydrogen from hydrogen sulfide by means of selective diffusionmembranes[J]. Int. J. Hydrogen Energy,1983,8(1):5-13.
[25]EDLUND D J,PLEDGER W A. Thermolysis of hydrogen sulfide in a metal-membrane reactor[J]. Journal of Membrane Science,1993,77(2/3):255-264.
[26]张谊华,滕玉美. 硫化铁催化剂的制备,表征及对H2S制H2反应的研究[J]. 华东理工大学学报(自然科学版),1995,21(6):738-742. ZHANG Y H,TENG Y M. The preparation and characterization of iron sulfide catalyst and it's reactivity for thermochemical decomposition of H2S to H2[J]. Journal of East China University of Science and Technology,1995,21(6):738-742.
[27]RESHETENKO T,KHAIRULIN S,ISMAGILOV Z,et al. Study of the reaction of high-temperature H2S decomposition on metal oxides(γ-Al2O3,α-Fe2O3,V2O5)[J]. Int. J. Hydrogen Energy,2002,27(4):387-394.
[28]RICARDO B V. Catalytic membrane reactor that is used for the decomposition of hydrogen sulphide into hydrogen and sulphur and the separation of the products of said decomposition:EP 1411029 [P]. 2004-04-21.
[29]STARTSEV A. Low-temperature catalytic decomposition of hydrogen sulfide into hydrogen and diatomic gaseous sulfur[J]. Kinet Catal.,2016,57(4):511-522.
[30]STARTSEV A,KRUGLYAKOVA O,CHESALOV Y A,et al. Low temperature catalytic decomposition of hydrogen sulfide into hydrogen and diatomic gaseous sulfur[J]. Top. Catal.,2013,56(11):969-980.
[31]STARTSEV A N,KRUGLYAKOVA O V. Diatomic gaseous sulfur obtained at low temperature catalytic decomposition of hydrogen sulfide [J]. Journal of Chemistry and Chemical Engineering,2013,7(11):1007-1013.
[32]STARTSEV A,KRUGLYAKOVA O,CHESALOV Y A,et al. Low-temperature catalytic decomposition of hydrogen sulfide on metal catalysts under layer of solvent[J]. Journal of Sulfur Chemistry,2016,37(2):229-240.
[33]BINGUE J P,SAVELIEV A V,FRIDMAN A,et al. Hydrogen sulfide filtration combustion:comparison of theory and experiment[J]. Experimental Thermal and Fluid Science,2002,26(2):409-415.
[34]SLIMANE R,LAU F,KHINKIS M,et al. Conversion of hydrogen sulfide to hydrogen by superadiabatic partial oxidation:thermodynamic consideration [J]. Int. J. Hydrogen Energy,2004,29(14):1471-1477.
[35]凌忠钱,周昊,钱欣平,等. 多孔介质内H2S贫氧燃烧制氢数值模拟[J]. 环境科学学报,2006,26(1):22-26. LING Z Q,ZHOU H,QIAN X P,et al. Numerical simulation of hydrogen production in porous media from hydrogen sulfide by partialoxidation[J]. Acta Scientiae Circumstantiae,2006,26(1):22-26.
[36]李国能,周昊,钱欣平,等. 多孔介质内H2S超绝热燃烧制氢的数值模拟[J]. 化工学报,2006,57(9):2175-2179. LI G N,ZHOU H,QIAN X P,et al. Modeling hydrogen production in super-adiabatic com bustion of hydrogen sulfide in porous media[J]. Journal of Chemical Industry and Engineering (China),2006,57(9):2175-2179.
[37]凌忠钱. 多孔介质内超绝热燃烧及硫化氢高温裂解制氢的试验研究和数值模拟[D]. 杭州:浙江大学,2008. LING Z Q. Experimental study and numerical simulation on super-adiabatic combustion and hydrogen production based on pyrolysis of H2S in porous media[D]. Hangzhou:Zhejiang University,2008.
[38]SHIH Y S,LEE J L. Continuous solvent extraction of sulfur from the electrochemical oxidation of a basic sulfide solution in the CSTER system [J]. Industrial & Engineering Chemistry Process Design and Development,1986,25(3):834-836.
[39]KALINA D W,MAAS JR E T. Indirect hydrogen sulfide conversion—Ⅱ. A basic electrochemical process [J]. Int. J. Hydrogen Energy,1985,10(3):163-167.
[40]罗文利,赵永丰. 从硫化氢中回收氢气和硫黄的方法[J]. 石油大学学报(自然科学版),1994,18(4):95-101. LUO W L,ZHAO Y F. A method for the recovery of hydrogen and element sulfur from hydrogen sulfide[J]. Journal of the University of Petroleum,China,1994,18(4):95-101.
[41]李发永,曹作刚,张海鹏,等. 由硫化氢制取硫黄及氢气扩大实验研究[J]. 化工进展,2001,20(7):38-41. Li F Y,CAO Z G,ZHANG H P,et al. Experimental study on transforming refinery acid tail gas into sulphur and hydrogen[J]. Chemical Industry and Engineering Progress,2001,20(7):38-41.
[42]袁长忠,邢定峰,俞英. 硫化氢间接电解制氢中试阴极动力学研究[J]. 化工学报,2005,56(7):1317-1321. YUAN C Z,XING D F,YU Y. Macroscopic kinetics of cathodic reaction in pilot- scale hydrogen production from indirect electrolysis of hydrogen sulfide[J]. Journal of Chemical Industry and Engineering(China),2005,56(7):1317-1321.
[43]SERPONE N,BORGARELLO E,GR TZEL M. Visible light induced generation of hydrogen from H2S in mixed semiconductor dispersions; improved efficiency through inter-particle electron transfer [J]. Journal of the Chemical Society,Chemical Communications,1984,6 :342-344.
[44]BORGARELLO E,SERPONE N,GR TZEL M,et al. Hydrogen production through microheterogeneous photocatalysis of hydrogen sulfide cleavage. The thiosulfate cycle [J]. Int. J. Hydrogen Energy,1985,10(11):737-741.
[45]KHAN M T,BHARDWAJ R,BHARDWAJ C. Photodecomposition of H2S by silver doped cadmium sulfide and mixed sulfides with ZnS[J]. Int. J. Hydrogen Energy,1988,13(1):7-10.
[46]DE G C,ROY A M,BHATTACHARYA S S. Photocatalytic production of hydrogen and concomitant cleavage of industrial waste hydrogen sulphide [J]. Int. J. Hydrogen Energy,1995,20(2):127-131.
[47]马贵军,鄢洪建,宗旭,等. 气-固相光催化分解硫化氢制氢[J]. 催化学报,2008,29(4):313-315. MA G J,YAN H J,ZONG X,et al. Photocatalytic splitting of H2S to produce hydrogen by gas solid phase reaction[J]. Chinese Journal of Catalysis,2008,29(4):313-315.
[48]SO W W,KIM K J,MOON S J. Photo-production of hydrogen over the CdS–TiO2nano-composite particulate films treated with TiCl4[J]. Int. J. Hydrogen Energy,2004,29(3):229-234.
[49]JANG J S,LI W,OH S H,et al. Fabrication of CdS/TiO2nano-bulk composite photocatalysts for hydrogen production from aqueous H2S solution under visible light[J]. Chemical Physics Letters,2006,425(4):278-282.
[50]RUBAN P,SELLAPPA K. Concurrent hydrogen production and hydrogen sulfide decomposition by solar photocatalysis[J].Clean——Soil,Air,Water,2016,44(9999):1-13.
[51]SUBRAMANIAN E,BAEG J O,KALE B B,et al. Visible light driven ZnFe2Ta2O9catalyzed decomposition of H2S for solar hydrogen production[J]. Bull. Korean Chem. Soc.,2007,28(11):2089-2092.
[52]KANADE K,BAEG J O,MULIK U,et al. Nano-CdS by polymer-inorganic solid-state reaction:visible light pristine photocatalyst for hydrogen generation[J]. Materials Research Bulletin,2006,41(12):2219-2225.
[53]WANG Z,CI X,DAI H,et al. One-step synthesis of highly active Ti-containing Cr-modified MCM-48 mesoporous material and the photocatalytic performance for decomposition of H2S under visible light[J]. Appl. Surf. Sci.,2012,258(20):8258-8263.
[54]KIM H H. Nonthermal plasma processing for air-pollution control:a historical review,current issues,and future prospects[J]. Plasma Processes and Polymers,2004,1(2):91-110.
[55]TRAUS I,SUHR H,HARRY J,et al. Application of a rotating high-pressure glow discharge for the dissociation of hydrogen sulfide[J]. Plasma Chemistry and Plasma Processing,1993,13(1):77-91.
[56]HELFRITCH D. Pulsed corona discharge for hydrogen sulfide decomposition[J]. IEEE Transactions on Industry Applications,1993,29(5):882-886.
[57]TRAUS I,SUHR H. Hydrogen sulfide dissociation in ozonizer discharges and operation of ozonizers at elevated temperatures [J]. Plasma Chemistry and Plasma Processing,1992,12(3):275-285.
[58]REDDY E L,KARUPPIAH J,SUBRAHMANYAM C. Kinetics of hydrogen sulfide decomposition in a DBD plasma reactor operated at high temperature [J]. Journal of Energy Chemistry,2013,22(3):382-386.
[59]REDDY E L,BIJU V,SUBRAHMANYAM C. Production of hydrogen from hydrogen sulfide assisted by dielectric barrier discharge [J]. Int. J. Hydrogen Energy,2012,37(3):2204-2209.
[60]REDDY E L,BIJU V,SUBRAHMANYAM C. Production of hydrogen and sulfur from hydrogen sulfide assisted by nonthermal plasma [J]. Applied Energy,2012,95:87-92.
[61]ZHAO L,WANG Y,JIN L,et al. Decomposition of hydrogen sulfide in non-thermal plasma aided by supported CdS and ZnS semiconductors [J]. Green Chemistry,2013,15(6):1509-1513.
[62]ZHAO L,WANG Y,LI X,et al. Hydrogen productionviadecomposition of hydrogen sulfide by synergy of non-thermal plasma and semiconductor catalysis[J]. Int. J. Hydrogen Energy,2013,38(34):14415-14423.
[63]RUSANOV V D,FRIDMAN A A. The physics of a chemically active plasma [J]. Moscow Izdatel Nauka,1984,1:416-429.
[64]FRIDMAN A. Plasma chemistry[M]. Oxford:Cambridge University Press,2008:738-749.
[65]DALAINE V,CORMIER J,PELLERIN S,et al. H2S destruction in 50 Hz and 25 kHz gliding arc reactors[J]. Journal of Applied Physics,1998,84(3):1215-1221.
[66]GUTSOL K,NUNNALLY T,RABINOVICH A,et al. Mechanisms of non-equilibrium dissociation of hydrogen sulfide in low-temperature plasma[C]//Proceedings of the 2010 IEEE International Conference on Plasma Science,Norfolk:IEEE,2010.
[67]KRASHENINNIKOV E,RUSANOV V,SANYUK S,et al. Dissociation of hydrogen sulfide in an RF discharge [J]. Soviet Physics——Technical Physics,1986,31(6):645-648.
[68]ASISOV R,VAKAR A,GUTSOL A,et al. Plasmachemical methods of energy carrier production[J]. Int. J. Hydrogen Energy,1985,10(7):475-477.
[69]BAGAUTDINOV A,ZHIVOTOV V,KALACHEV I,et al. Dissociation of hydrogen sulfide in a mixture with carbon dioxide gas in a high-power microwave discharge[J]. Soviet Physics——Technical Physics,1991,36(4):488-490.
[70]BAGAUTDINOV A,JIVOTOV V,EREMENKO J,et al. Plasma chemical production of hydrogen from H2S-containing gases in MCW discharge[J]. Int. J. Hydrogen Energy,1995,20(3):193-195.
[71]BAGAUTDINOV A,ZHIVOTOV V,KALACHEV I,et al. Investigations of the radial distributions of gas-flows in a high-power microwave-discharge[J]. High Energy Chemistry,1993,27(4):305-310.
[72]BAGAUTDINOV A,JIVOTOV V,EREMENKO Y,et al. Plasmachemical hydrogen production from natural gases containing hydrogen sulfide [J]. Hydrogen Energy Progress,1998,1:683-690.
[73]COX B G,CLARKE P F,PRUDEN B B. Economics of thermal dissociation of H2S to produce hydrogen[J]. Int. J. Hydrogen Energy,1998,23(7):531-544.
[74]董永治,王涵慧,俞稼镛. 微波等离子体方法分解H2S制氢[J]. 太阳能学报,1997,18(2):142-145. DONG Y Z,WANG H H,YU J Y. Hydrogen production by H2S microwave plasma dissociation[J]. Acta Energiae Solaris Sinica,1997,18(2):142-145.
[75]汪建华,徐尧,高建保,等. 常压微波等离子体微波功率对硫化氢分解效率的影响[J]. 武汉工程大学学报,2013,35(3):34-37. WANG J H,XU Y,GAO J B,et al. Influence of microwave power on decomposition of hydrogen sulfide by atmospheric microwave plasma[J]. Journal of Wuhan Institute of Technology,2013,35(3):34-37.
[76]徐尧,汪建华,高建保,等. 大气微波等离子体射流处理H2S废气[J]. 强激光与粒子束,2013,25(11):2909-2913. XU Y,WANG J H,GAO J B,et al. Treatment of waste H2S by atomospheric pressure microwave plasma jet[J]. High Power Laser and Particle Beams,2013,25(11):2909-2913.
Research progress on hydrogen and sulfur production from direct decomposition of hydrogen sulfide
ZHANG Jing,ZHANG Tie,SUN Feng,XU Wei,SHI Ning
(State Key Laboratory of Safety and Control for Chemicals,SINOPEC Research Institute of Safety Engineering,Qingdao 266071,Shandong,China)
Direct decomposition of hydrogen sulfide is an effective way not only to treat the waste gas in coal,petroleum,natural gas and mineral processing industries for solving the environmental pollution problems but also to produce sulfur and a clean energy,hydrogen. In this paper,the research progress of hydrogen and sulfur produced from direct decomposition of hydrogen sulfide including themolysis(directly high–temperature thermolysis,catalytic thermolysis and superadiabatic thermolysis),electrochemical technology,photocatalytic decomposition and plasma technology are reviewed. Fundamental principle and themodynamic basis of methods involved are briefly introduced. Then, a detailed description of research status focused on research methods,technique feature,reaction performance,advantage and disadvantage,and potential breakthrough point,etc. is given. The future development trend is prospected at the end of this review as well. A prospect is put forward that continuously exploring and developting new technologies such as combining membrane technology,catalytic technology and plasma technology would be the future trend in the field of hydrogen production from H2S decomposition.
hydrogen production;pyrolysis;recovery
TQ125.1
A
1000–6613(2017)04–1448–12
10.16085/j.issn.1000-6613.2017.04.039
2016-08-10;修改稿日期:2016-12-21。
国家自然科学基金项目(21503279)。
及联系人:张婧(1986—),女,博士,博士后,主要从事等离子体化学及等离子体-催化化学反应研究。E-mail:jingdlut@foxmail.com。
猜你喜欢
化工管理(2022年14期)2022-12-02
陶瓷学报(2021年5期)2021-11-22
中国特种设备安全(2020年11期)2020-06-09
上海建材(2020年12期)2020-04-13
陶瓷学报(2019年6期)2019-10-27
中成药(2018年9期)2018-10-09
通信电源技术(2018年5期)2018-08-23
浙江农业科学(2016年11期)2016-05-04
橡胶工业(2015年10期)2015-08-01
橡胶工业(2015年4期)2015-07-29
