
比卡鲁胺的合成工艺改进
2017-04-27 03:13李云龙冯菊红葛燕丽胡学雷
化学与生物工程 2017年4期
李云龙,冯菊红*,葛燕丽,胡学雷
(1.武汉工程大学化工与制药学院,湖北 武汉 430074;2.绿色化工过程教育部重点实验室,湖北 武汉 430074)
比卡鲁胺的合成工艺改进
李云龙1,2,冯菊红1,2*,葛燕丽1,2,胡学雷1,2
(1.武汉工程大学化工与制药学院,湖北 武汉 430074;2.绿色化工过程教育部重点实验室,湖北 武汉 430074)
以甲基丙烯酰氯为原料,经N-酰化、环氧化、缩合和氧化合成了比卡鲁胺,总收率为57.6%,纯度为98.3%,并通过1HNMR、MS对其结构进行了确认。
比卡鲁胺;药物合成;工艺改进
比卡鲁胺(bicalutamide),化学名为(±)-N-[4-氰基-3-(三氟甲基)苯基]-3-[(4-氟苯基)磺酰基]-2-羟基-2-甲基丙酰胺,是捷利康公司开发的一种非甾体类抗雄激素药物,商品名为康士得(Casodex)。比卡鲁胺一般与促黄体激素释放激素类似物[如亮丙瑞林(Leuprorelin)、戈舍瑞林(Goserelin)]等联合使用,非常适于晚期前列腺癌的治疗[1],其作用特异性强,口服有效,给药方便,耐受性好,半衰期较长[2],疗效较好,在医药市场供不应求。比卡鲁胺的合成路线较多,可以溴丙酮、甲基丙烯酰氯、甲基丙烯酸甲酯、丙酮酸、3-溴-2-甲基-2-羟基丙酸或2-甲基丙烯酰胺为原料合成[3-10]。这些方法或多或少存在原料昂贵、反应时间长、操作复杂、收率低、不安全、不环保等缺点,因此,开发高效、环保、适于工业化的比卡鲁胺合成方法尤为重要。
作者以甲基丙烯酰氯(Ⅱ)为原料,经与4-氨基-2-三氟甲基苯甲腈(Ⅲ)N-酰化生成N-[4-氰基-3-(三氟甲基)苯基]-2-甲基丙烯酰胺(Ⅳ),碳碳双键环氧化生成N-[4-氰基-3-(三氟甲基)苯基]-2-甲基-2-环氧丙酰胺(Ⅴ),再与对氟苯硫酚缩合得N-[4-氰基-3-(三氟甲基)苯基]-3-[(4-氟苯基)硫]-2-羟基-2-甲基丙酰胺(Ⅵ),最后氧化得到目标化合物比卡鲁胺(Ⅰ)。合成路线如图1所示。
1 实验
1.1 试剂与仪器
邻溴三氟甲苯(质量分数99%),山东西亚化学工业有限公司;甲基丙烯酰氯(质量分数95%)、对氟苯硫酚(质量分数98%),阿拉丁试剂公司;其它试剂均为分析纯。
API-150EXLC/MS型高分辨质谱仪;BrukerARX400型核磁共振仪,氘带氯仿或氘带二甲基亚砜为溶剂,TMS为内标;RY-1G型熔点仪,天津天光光学仪器有限公司。

图1 比卡鲁胺的合成路线Fig.1 Synthetic route of bicalutamide
1.2 方法
1.2.1 中间体Ⅲ的的合成
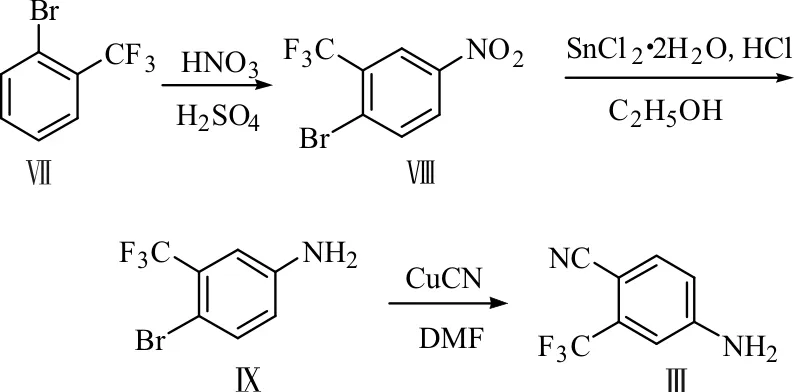
(1)2-溴-5-硝基三氟甲苯(Ⅷ)的合成
在三口烧瓶中加入邻溴三氟甲苯(Ⅶ)(20.0 g,88.9 mmol),冰浴冷却至15 ℃,搅拌下滴加浓硝酸(7.7 mL)和浓硫酸(23.1 mL)混合物,控制反应温度在25 ℃以下,1 h滴加完毕,再于室温下反应3 h。将反应液移至碎冰(80 g)中静置1 h,析出淡黄色固体,过滤并用冰水冲洗直至pH值为中性,用乙醇溶液(95%)重结晶,得类白色固体化合物Ⅷ(18.5 g)。
(2)3-三氟甲基-4-溴苯胺(Ⅸ)的合成
在三口烧瓶中加入二水合氯化亚锡(33.8 g,150.0 mmol)、浓盐酸(32.2 mL)和无水乙醇(33.8 mL),搅拌至澄清,加热至37 ℃,搅拌下滴加化合物Ⅷ(15.0 g,55.6 mmol)和无水乙醇(30.1 mL)的混合物,0.5 h滴加完毕,在40~45 ℃反应3 h。用氢氧化钠溶液调pH值至弱碱性成乳浊状,将乳浊液过滤,氯仿萃取,减压蒸馏得淡黄色澄清溶液,加适量石油醚析出淡黄色固体,过滤并用石油醚冲洗,所得固体用石油醚重结晶,得白色固体化合物Ⅸ(11.7 g)。
(3)目标中间体Ⅲ的合成
在三口烧瓶中加入化合物Ⅸ(10 g,41.7 mmol)和DMF(50 mL),搅拌下加入氰化亚铜(4.5 g,50.0 mmol),160 ℃回流反应2.5 h,降温倒入水中,并用乙酸乙酯萃取,然后分别用水和饱和食盐水洗涤,干燥,减压蒸馏得棕黑色固体,再用乙醇溶液(50%和30%)重结晶,得棕色固体即目标中间体Ⅲ(3.8 g)。
1.2.2 化合物Ⅳ的合成
在三口烧瓶中加入中间体Ⅲ(3.6 g,19.4 mmol)、三乙胺(5.8 g,57.8 mmol)和二氯甲烷(28.8 mL),控制温度在-5 ℃左右搅拌0.5 h,滴加化合物Ⅱ(2.0 g,22.3 mmol)和二氯甲烷(7.0 mL)的混合物,1 h滴加完毕,并在0 ℃左右反应3 h。向反应液中加入水,用二氯甲烷萃取,分别用饱和碳酸氢钠溶液、饱和氯化钠溶液和水洗涤,干燥,减压蒸馏得黄色固体,再用甲苯重结晶,得类白色固体化合物Ⅳ(4.2 g)。
1.2.3 化合物Ⅴ的合成
在三口烧瓶中加入化合物Ⅳ(3.0 g,11.8 mmol),用甲酸(22.5 mL)溶解,室温下搅拌0.5 h,缓慢滴加过氧化氢(12 mL,30%),1 h滴加完毕,反应2 h。将反应液用二氯甲烷萃取,分别用饱和连二亚硫酸钠溶液、饱和碳酸氢钠溶液和饱和氯化钠溶液洗涤,干燥,减压蒸馏得黄色固体,再用无水乙醇重结晶,得淡黄色固体化合物Ⅴ(2.5 g)。
1.2.4 化合物Ⅵ的合成
在三口烧瓶中加入对氟苯硫酚(2.0 g,15.7 mmol)和甲醇(4 mL),冷却至0 ℃,滴加氢氧化钠溶液(1 mL,50%),倒入化合物Ⅴ(2.0 g,7.4 mmol),继续搅拌1.5 h。将反应液用乙醚萃取,用浓盐酸调pH值小于7,用饱和氯化钠溶液洗涤,干燥,减压蒸馏得淡黄色黏稠物,再用石油醚-甲苯(5∶1,体积比)重结晶,得淡黄色固体化合物Ⅵ(2.6 g)。
1.2.5 比卡鲁胺(Ⅰ)的合成
在三口烧瓶中加入化合物Ⅵ(2.0 g,5.0 mmol)、丙酮(20 mL)、二水合钨酸钠(0.25 g)和甲酸(5 mL),搅拌0.5 h,滴加过氧化氢(5 mL,30%)后继续搅拌1 h,将反应液用二氯甲烷萃取,分别用饱和连二亚硫酸钠溶液、饱和碳酸氢钠溶液和饱和氯化钠溶液洗涤,干燥,减压蒸馏得类白色固体,再用无水乙醇重结晶,得白色固体化合物 Ⅰ(2.1 g),即目标化合物比卡鲁胺。
2 结果与讨论
2.1 中间体与目标化合物的表征
化合物Ⅷ:收率77.1%, m.p.45~47 ℃(文献值46~48 ℃[11])。1HNMR(CDCl3),δ:8.57~8.56(d,J=2.5 Hz,1H),8.28~8.25(dd,J=8.6 Hz,2.8 Hz,1H),7.96~7.94(d,J=8.7 Hz,1H) 。
化合物Ⅸ:收率88.0%,m.p.52~54 ℃(文献值55~56 ℃[11])。 ESI-MS,m/z:241.8 [M+H]+;1HNMR(CDCl3),δ:7.42~7.40(d,J=8.4 Hz,1H),6.98~6.97(d,J=2.8 Hz,1H),6.67~6.65(dd,J=8.4 Hz,2.8 Hz,1H),3.86(s,2H)。
化合物Ⅲ:收率49%,m.p.140~142 ℃(文献值142~143 ℃[11])。ESI-MS,m/z:187.3 [M+H]+;1HNMR(CDCl3),δ:7.58~7.56(d,J=8.7 Hz,1H),6.95(d,J=2.2 Hz,1H),6.80~6.77(dd,J=8.4 Hz,2.2 Hz,1H),4.39(s,2H) 。
化合物Ⅳ:收率85.7%,m.p.135~137 ℃(文献值137~139 ℃[12])。ESI-MS,m/z:255.2 [M+H]+;1HNMR(CDCl3),δ:8.06~8.05(d,J=2.0 Hz,1H),7.99~7.97(dd,J=8.4 Hz,2.0 Hz,1H),7.89(s,1H),7.82~7.79(d,J=8.4 Hz,1H),5.87(s,1H),5.61(d,J=1.4 Hz,1H),2.08(s,3H) 。
化合物Ⅴ:收率78.4%,m.p.147~149 ℃(文献值149~150 ℃[13])。 ESI-MS,m/z:271.1 [M+H]+;1HNMR(CDCl3),δ:8.41(s,1H),8.02(d,J=2.0 Hz,1H),7.92~7.89(dd,J=8.4 Hz,2.0 Hz,1H),7.81~7.79(d,J=8.4 Hz,1H),3.01(s,2H),1.69(s,3H)。
化合物Ⅵ:收率88.2%,m.p.114~115 ℃(文献值116~117 ℃[6])。ESI-MS,m/z:399.1 [M+H]+;1HNMR(CDCl3),δ:8.99(s,1H),7.91(d,J=1.4 Hz,1H),7.78~7.72(m,2H),7.41~7.38(dd,J=9.0 Hz,5.0 Hz,2H),6.90~6.86(t,J=17.1 Hz,2H),3.79~3.75(d,J=14.3 Hz,1H),3.56(s,1H),3.11~3.08(d,J=14.3 Hz,1H),1.53(s,3H)。
目标化合物Ⅰ:收率97.2%,m.p.189~190 ℃(文献值191~193 ℃[6]);纯度98.3%[HPLC归一化法:色谱柱 Dionex 6040.2365 Viper Cap(0.18 mm×450 mm);流动相为甲醇-水(60∶40,体积比);检测波长270 nm;柱温15 ℃;流速1.0 mL·min-1]。ESI-MS,m/z:431.1 [M+H]+;1HNMR(DMSO-d6),δ:10.40(s,1H),8.45~8.44(d,J=2.0 Hz,1H),8.24~8.22(dd,J=8.7 Hz,2.0 Hz,1H),8.11~8.09(d,J=8.7 Hz,1H),7.95~7.92(dd,J=9.0 Hz,5.0 Hz,2H),7.41~7.36(t,J=17.6 Hz,2H),6.43(s,1H),3.97~3.94(d,J=14.9 Hz,1H),3.75~3.71(d,J=14.9 Hz,1H),1.41(s,3H)。
2.2 合成方法改进
(1)合成化合物Ⅸ时,文献[11]采用水蒸气蒸馏法,用时长、操作繁琐且不安全,收率为76.0%。本合成路线采用砂芯漏斗抽滤乳浊液,再进行萃取的方法,用时短、操作简单,收率提高到88.0%。
(2) 合成化合物Ⅳ时,文献[6]将化合物Ⅲ滴加到化合物Ⅱ中,但是由于N-酰化是个比较活泼的放热反应,容易发生双取代,所以这种滴加方式会产生较多的双取代副产物,收率为78.7%。本合成路线将滴加次序反过来,将化合物Ⅱ滴加到化合物Ⅲ中,且在滴加时,先用溶剂将化合物Ⅱ稀释适当倍数,以控制反应速率,收率提高到85.7%。
(3) 合成化合物Ⅴ时,文献[6]使用的氧化剂是危险易爆的间氯过氧化苯甲酸(m-CPBA),且其副产物间氯苯甲酸对环境非常有害,收率为65.0%。本合成路线使用安全的30%过氧化氢和甲酸作氧化剂,副产物为水,收率提高到78.4%。这个改进同样应用于目标化合物Ⅰ的合成,在氧化化合物Ⅵ的时候,使用30%过氧化氢、甲酸和二水合钨酸钠作氧化剂代替文献中的m-CPBA,安全环保,反应温和,收率较高。
3 结论
以甲基丙烯酰氯为原料,经N-酰化、环氧化、缩合和氧化合成了比卡鲁胺,总收率为57.6%,纯度为98.3%,并通过1HNMR、MS对其结构进行了确认。通过简化后处理、调整反应物滴加顺序、控制浓度、优化氧化剂,将总收率提高到57.6%,该合成方法绿色环保,条件温和,更适于工业化生产。
[1] SEPP-LORENZINO L,SLOVIN S.Prostate cancer:therapeutic patent review[J].Expert Opinion on Therapeutic Patents,2000,10(12):1833-1842.
[2] 李艳芹,诸慧,倪旭晖,等.比卡鲁胺相关物质B的合成和表征及理化性质[J].中国现代应用药学,2013,30(8):847-850.
[3] NAIR V A,MUSTAFA S M,MOHLER M L,et al.Synthesis of novel iodo derived bicalutamide analogs[J].Tetrahedron Letters,2004,45(51):9475-9477.
[4] TETSUYA S,TADASHI K,KIYOSHI S,et al.Process for producing bicalutamide and method of purifying intermediate therefor:WO 2005037777[P].2005-04-28.
[5] BOR A,OROSZ G,LUKACS F,et al.Method for producing bicalutamide:US 2005033082[P].2005-02-10.
[6] 刘雅茹,冯雪松,孟繁浩,等.抗雄激素药物比卡鲁胺的合成[J].中国医科大学学报,2005,34(6):518-519.
[7] 柴洪伟,奚祯浩,金健,等.N-[4-氰基-3-(三氟甲基)苯基]-2-甲基-2-环氧丙酰胺合成工艺改进研究[J].精细化工中间体,2014,44(5):43-45.
[8] 李艳芹,倪旭晖.比卡鲁胺的合成[J].浙江化工,2013,44(6):7-9.
[9] 肖涛,张孝清,田春梅,等.比卡鲁胺的合成[J].合成化学,2003,11(4):346-348.
[10] EKWURIBE N N,JAMES K D.Methods of synthesizing acylanilides including bicalutamide and derivatives thereof:US 2003045742[P].2003-03-06.
[11] 刘雅茹,巴琳,冯雪松,等.3-三氟甲基-4-氰基苯胺的合成[J].广东药学院学报,2005,21(3):243-244.
[12] THIJS L.Process for making bicalutamide and intermediates the-reof:US 20040068135[P].2004-04-08.
[13] CHEN B C,ZHAO R,GOVE S,et al.Nucleophilic aromatic substitution of methacrylamide anion and its application to the synthesis of the anticancer drug bicalutamide[J].Journal of Organic Chemistry,2003,68(26):10181-10182.
Improvement on Synthetic Process of Bicalutamide
LI Yun-long1,2,FENG Ju-hong1,2*,GE Yan-li1,2,HU Xue-lei1,2
(1.SchoolofChemicalEngineering&Pharmacy,WuhanInstituteofTechnology,Wuhan430074,China;2.KeyLaboratoryofGreenChemicalProcessofMinistryofEducation,Wuhan430074,China)
Usingmethacryloylchlorideasarawmaterial,bicalutamidewiththeoverallyieldof57.6%andthepurityof98.3%wassynthesizedbyafour-stepreactionofN-acylation,epoxidation,condensationandoxidation.Thestructureofbicalutamidepreparedwasconfirmedby1HNMRandMS.
bicalutamide;drugsynthesis;processimprovement
湖北省自然科学基金重点项目(2011CDA048),武汉工程大学研究生创新基金资助项目(CX2015074)
2016-12-08
李云龙(1992-),男,湖北仙桃人,硕士研究生,研究方向:药物合成,E-mail:1210839413@qq.com;通讯作者:冯菊红,博士,讲师,E-mail:jhfeng@wit.edu.cn。
10.3969/j.issn.1672-5425.2017.04.009
TQ463.4 R914.5
A
1672-5425(2017)04-0039-03
李云龙,冯菊红,葛燕丽,等.比卡鲁胺的合成工艺改进[J].化学与生物工程,2017,34(4):39-41,46.
猜你喜欢
浙江化工(2022年8期)2022-09-05
有机氟工业(2020年2期)2020-07-04
农药科学与管理(2019年8期)2019-11-23
益寿宝典(2017年1期)2017-09-03
中小学实验与装备(2016年1期)2016-04-19
浙江化工(2015年4期)2015-11-28
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
红领巾·探索(2014年8期)2014-10-10
江西理工大学学报(2013年1期)2013-03-20
郑州大学学报(理学版)(2013年3期)2013-03-11
