超滤亲和结合液相色谱—质谱联用和分子对接技术筛选毛菊苣种子中高亲和性α—葡萄糖苷酶抑制剂
2017-06-15 17:21陈海君秦惠玉龙飞于玮王颖慧陈露
分析化学 2017年6期
陈海君+秦惠玉+龙飞+于玮+王颖慧+陈露君+李全凯+陈文+秦冬梅+韩博
摘 要 采用超滤亲和结合液相色谱质谱联用(UFLCMS) 和分子对接技术筛选毛菊苣种子中高亲和α葡萄糖苷酶抑制剂。以4硝基苯αD吡喃葡萄糖苷(PNPG)为底物,阿卡波糖为阳性对照,评价毛菊苣种子提取物对α葡萄糖苷酶的抑制活性,其中阿卡波糖IC50为0.003 mg/mL,毛菊苣种子IC50为0.447 mg/mL。利用UFLCMS技术对毛菊苣种子提取物进行筛选鉴定,获得4种化合物; 通过Autodock软件筛选出2种与α葡萄糖苷酶有较高亲和力的化合物,分别是绿原酸和异绿原酸A。结合体外酶活实验,验证了绿原酸、异绿原酸A对α葡萄糖苷酶的抑制活性。结果表明,各化合物对α葡萄糖苷酶的抑制活性由大到小依次是:阿卡波糖>异绿原酸A>绿原酸,其中异绿原酸A与阿卡波糖抑制率相近。
关键词 超滤亲和; 液相色谱质谱联用; 分子对接; 毛菊苣种子; α葡萄糖苷酶抑制剂
1 引 言
目前,从天然产物中发现2型糖尿病(Type 2 diabetes mellitus,T2DM) [1,2]治疗药物是治疗糖尿病的研究热点之一。α葡萄糖苷酶抑制剂已被广泛用于治疗T2DM,拜唐苹作为α葡萄糖苷酶抑制剂的代表药物,主要成分为阿卡波糖,其疗效已被临床研究和应用所证实。
毛菊苣(Cichorium glandulosum Boiss. et Hout) 為菊科菊苣属植物[3,4],是维吾尔族医生习用药材,常以地上部分入药,具清肝利胆、健胃消食、利尿消肿之功效[5,6],被中华人民共和国卫生计生委药食同源药材目录(2015版) 列为“药食同源类”药材。研究发现,毛菊苣具有良好的降糖活性[7],但目前研究主要集中于地上部分,其种子部位降糖物质基础尚不明确。探索毛菊苣种子(Cichorium glandulosum Boiss. et Hout seed, CGS) 的作用机制,寻找其降糖的物质基础是深入开发毛菊苣种子的关键。
传统中药分离方法主要为柱层析、两相溶剂萃取法、系统溶剂分离法等[8,9]。但天然产物组成复杂,分离纯化耗时长,操作复杂[10]。近年来建立的超滤亲和液相色谱质谱联用(Ultra filtration affinityliquid chromatographymass spectrometry,UFLCMS) 技术可在很大程度上克服传统方法的缺点,在药物筛选中显示了良好的实用性和高效性[11],但采用该技术时仍存在假阳性结果。研究者开发了多种方法减少假阳性结果, 如明胶沉淀法结合在线筛选法[12,13]、多组实验对照法[14, 15]、键和磁珠筛选法[16~18]。
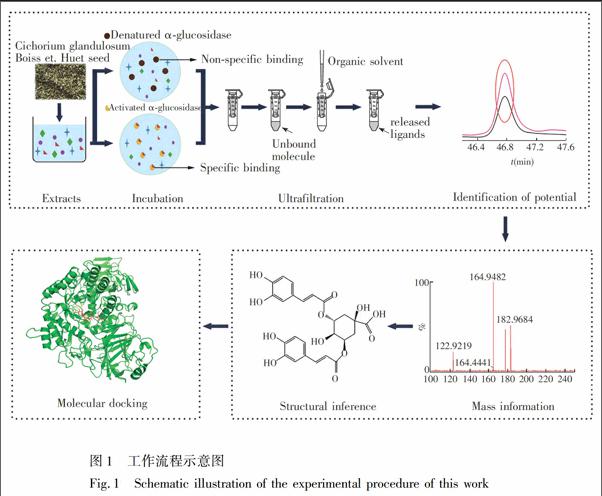
为提高筛选质量,本研究对UFLCMS筛选技术进行改进和优化,结合分子对接技术(Molecular docking) 筛选毛菊苣种子中高亲和性的α葡萄糖苷酶抑制剂,为毛菊苣种子的降糖活性物质基础研究提供了参考。本研究的工作流程图如图1所示。
2 实验部分
2.1 仪器与试剂
Waters2545型高效液相色谱仪(配2489紫外检测器) 、Xevo TQD质谱联用仪(美国Waters公司) ; 3001型多功能酶标仪(美国热电公司) ; Microcon超滤离心管(YM10,MWCO 10 KDa, 美国Millipore公司) ; Centrifuge5424R高速冷冻离心机(德国Eppendorf公司) ; QL861涡旋混合器(海门市其林贝尔仪器制造有限公司) ; MiliQ Advantage A10超纯水系统(美国Millipore公司) 。
α葡萄糖苷酶(50~120KD) 、4硝基苯αD吡喃葡萄糖苷(PNPG) 、阿卡波糖(SigmaAldrich公司) ; 绿原酸、异绿原酸A对照品(上海源叶生物科技有限公司) ; 甲醇和乙腈(色谱纯,美国Thermo Fisher公司) ; 其余试剂为分析纯,实验用水为MilliQ超纯水。
毛菊苣种子购于新疆维吾尔自治区和田县,经石河子大学药学院韩博副教授鉴定系毛菊苣种子,存放于新疆特种植物药重点实验室药剂学研究室。
2.2 毛菊苣种子样品处理
称取毛菊苣种子约30 g, 用95%乙醇回流提取3次,每次2.5 h,合并提取液,稀释至4 mg/mL。将α葡萄糖苷酶冻用10 mmol/L 磷酸盐缓冲溶液(PBS,pH 6.86)配制成2 U/mL的溶液,用于酶活性分析。
2.3 毛菊苣种子提取物的α葡萄糖苷酶活性抑制实验
参照文献[19,20]方法,以PNPG为底物,阿卡波糖为阳性对照,测定毛菊苣种子提取物的酶抑制活性。实验分为空白组、阳性对照组、样品组和背景组(n=3) 。依次加入PBS缓冲液、样品和α葡萄糖苷酶液,于37℃反应15 min,加入PNPG底物溶液,孵育20 min,加入0.2 mol/L Na2CO3溶液终止反应,立即测定405 nm处吸光度值。
2.5 HPLCMS/MS分析条件
HPLC条件:Symmetry C18分析柱(250 mm × 4.6 mm, 5 μm) ; 柱温25℃; 梯度洗脱:流动相A为0.2%甲酸溶液,B为乙腈; 梯度洗脱:0~5 min,12% B; 5~20 min,12%~25% B; 20~45 min,25%~40%B; 45~60 min,40%~50% B; 流速1.0 mL/min; 检测波长254 nm; 进样量20 μL。
电喷雾质谱(ESIMS) 条件:正、负离子电离模式,扫描范围m/z 100~2000 Da,实验前质量数均经过校正,毛细管电压2 kV。ESI电喷雾离子源温度200℃,锥孔气为氮气,碰撞气为氦气,锥孔电压30~60 kV,碰撞电压15~30 V。
2.6 分子对接
利用Autodock vina软件(Scripps研究所的Olson实验室) 进行分子对接,在NCBI蛋白质数据库中用PSIBLAST在Protein Data Bank数据库进行序列相似性搜索。使用PYMOL 1.2(DeLanoScientific LLC公司) 软件作图。
选取α葡萄糖苷酶晶体(PDB ID: 2QMJ) ,在上述构建好的α葡萄糖苷酶的三维结构基础上,利用Autodock tools(Scripps研究所的Olson实验室) 剥离蛋白质三维结构中的溶剂水分子和调节原子,根据配体结构特性找到结合部位,将存储文件中的化合物与蛋白质分子进行柔性对接,在每一个对接后,移动小分子的位置,使之最匹配。排除结合程度较低、位置不佳的化合物,记录配体与受体对接位置及计算评分。以上所用参数除特别指明外,均为默认参数。
2.7 筛选得到的化合物的抑制活性验证
参照2.4节方法,对毛菊苣种子中经UFLCMS和分子对接筛选出的化合物进行体外酶活性抑制实验的验证。
3 结果与讨论
3.1 毛菊苣种子提取物对α葡萄糖苷酶的抑制活性
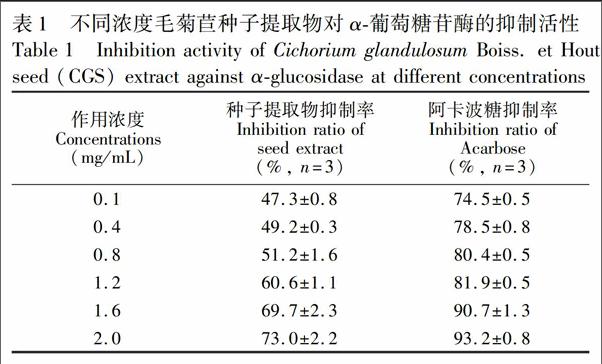
如表1所示, 2.0 mg/mL阿卡波糖对α葡萄糖苷酶的抑制率为93.2%,IC50為0.003 mg/mL。相同浓度的毛菊苣种子提取物对α葡萄糖苷酶的抑制率为73.0%,IC50为0.447 mg/mL,表明毛菊苣种子具有良好的α葡萄糖苷酶抑制活性,可作为进一步复筛的对象。
3.2 毛菊苣种子提取物的超滤亲和液相色谱质谱联用分析
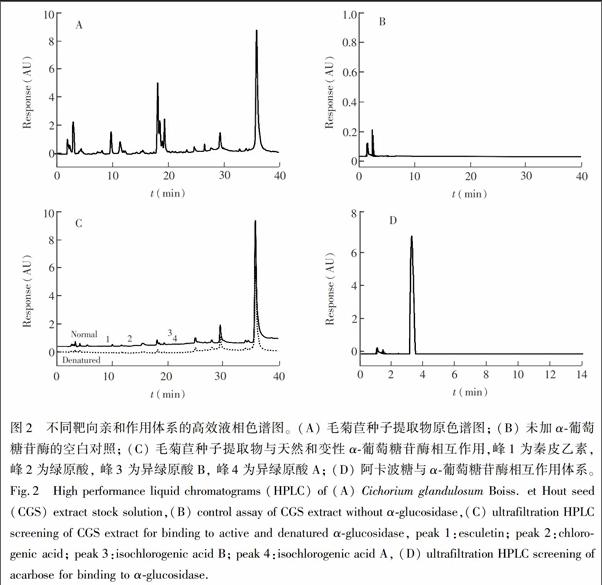
按照2.4节的方法,筛选毛菊苣种子提取物中与α葡萄糖苷酶特异性结合的小分子。与毛菊苣种子提取物的色谱图(图2A) 相比发现,在超滤筛选实验条件下,毛菊苣种子提取物中只有部分化合物能够与α葡萄糖苷酶结合(图2C) ; 与空白对照的色谱图(图2B)相比发现,本方法可有效避免小分子化合物吸附到超滤膜而造成的假阳性结果。
天然酶捕获的配体所对应的峰面积大于其灭活酶对照的峰面积。因此峰面积的变化情况可反映化合物与α葡萄糖苷酶的结合率,图2C中未标注序号的峰的峰高和峰面积无显著性差异,这可能由于浓度较大的化合物占据α葡萄糖苷酶活性位点的概率比浓度小的化合物大所致。通过对比结合率,筛选出4个峰(图2C) ,各峰与α葡萄糖苷酶的结合率均大于19.0%,表明毛菊苣种子提取物中的活性成分能够与α葡萄糖苷酶特异性结合。以阳性对照阿卡波糖进行超滤实验(图2D),表明本方法准确性较好。
3.3 液相色谱质谱联用分析和结构鉴定
根据超滤亲和筛选出的4种化合物的保留时间、准分子离子峰和二级质谱碎片信息(表2) ,结合参考文献进行解析,结果见图3和图4。紫外光谱分析结果(图3A、3B; 图4A、4B) 表明,化合物2,3和4具有相似的紫外吸收行为,最大吸收峰在325 nm附近; 而化合物1的最大吸收峰在330 nm处。化合物1准分子离子峰为m/z 177,在二级质谱分析中丢失两分子CO,产生碎片离子为m/z 151、123和105,具有较高的相对丰度,结合保留时间和文献[5],鉴定化合物1为秦皮乙素。化合物1在负离子模式下的一级质谱图和二级质谱图如图3C和图3E所示。
化合物2准分子离子峰为m/z 353,在二级质谱分析中丢失一分子C6H10O6,产生碎片离子m/z 183,继而丢失一分子H2O,产生碎片离子m/z 165,结合保留时间和文献[24,25],鉴定化合物2为绿原酸。化合物2在负离子模式下的一级质谱图和二级质谱图如图3D和图3F所示。
化合物3准分子离子峰为m/z 515,在二级质谱分析产生碎片离子m/z 353、179和135,具有较高的相对丰度。结合异绿原酸B保留时间和文献[26],鉴定化合物3为异绿原酸B。化合物3在负离子模式下的一级质谱图和二级质谱图如图4C和图4E所示。
化合物4准分子离子峰为m/z 515,在二级质谱分析产生碎片离子m/z 353和191,具有较高的相对丰度。结合异绿原酸A保留时间和文献[26],鉴定化合物4为异绿原酸A。化合物4在负离子模式下的一级质谱图和二级质谱图如图4D和图4F所示。
3.4 分子对接结果
以阿卡波糖为阳性对照,采用分子对接方法,评价对接评分和空间结合模式,结果见图5。对接评分次序为:异绿原酸A>绿原酸>异绿原酸B>秦皮乙素。从空腔位置分析,毛菊苣种子化合物中秦皮乙素、异绿原酸B可与α葡萄糖苷酶结合,但空腔位置占据较小,缺乏氢键支持。绿原酸和异绿原酸A空腔占据位置较大,氢键数多,推测绿原酸和异绿原酸A可作为高亲和力的α葡萄糖抑制剂。
3.5 活性实验验证结果
根据UFLCMS和分子对接筛选结果,对毛菊苣种子中筛选出潜在的α葡萄糖苷酶抑制剂绿原酸和异绿原酸A进行了体外酶活性抑制实验,结果见表3。体外酶活性抑制实验结果表明,绿原酸和异绿原酸A对α葡萄糖苷酶的抑制活性较强,与分子对接筛选结果吻合。抑制率顺序为阿卡波糖>异绿原酸A>绿原酸,且各化合物都呈现明显的浓度依赖性。
4 结 论
通过建立酶抑制剂模型,发现毛菊苣种子提取物具有良好的α葡萄糖苷酶抑制活性。利用UFLCMS技术从毛菊苣种子提取物中筛选并鉴定出4种潜在的α葡萄糖苷酶抑制剂,初步鉴定为为秦皮乙素、绿原酸、异绿原酸A和异绿原酸B。通过分子对接技术,在常规的对接得分评价匹配性的基础上,分析4种化合物与α葡萄糖苷酶结合程度和对接评分,得到异绿原酸A和绿原酸两种高亲和力的酶抑制剂,并进行酶活性验证。结果表明,随着作用浓度提高,其抑制率逐渐提高,并呈现出一定的浓度依赖性,异绿原酸A的酶活性抑制率大于绿原酸。文献[27~31]表明,这两种化合物具降糖功效,与本研究结果吻合。本研究结果为开发基于毛菊苣的降血糖产品提供了依据,也为在天然产物中发现高亲和力的酶抑制剂提供了方法。
References
1 Trinh B T, Staerk D, Jager A K. J. Ethnopharmacol., 2016, 186: 189-195
2 Yang D, Zhao J, Liu S, Song F, Liu Z. Anal. Methods, 2014, 6(10): 3353-3359
3 Upur H, Amat N, Blaekovic' B, Talip A. Food Chem. Toxicol., 2009, 47(8): 2022-2030
4 ZHANG Yao, XIN Xuelei, Haji Akber AISA. Chinese Traditional Patent Medicine, 2012, 34(12): 2322-2325
张 尧, 信学雷, 阿吉艾克拜尔·艾萨. 中成药, 2012, 34(12): 2322-2325
5 Yang W Z , Hao W, Jing S, Feng F. Chinese J. Nat. Med., 2009, 7(3): 193-195
6 Ding L, Liu J L, Hassan W, Wang L L, Yan F R, Shang J. Sci. Rep., 2014, 4(17): 4715
7 XIN XueLei, WU HanKui, LYU QiaoYing, HAJI Akber Aisa. Nat. Prod. Res. Dev., 2012, 24(2): 234-238
信學雷, 吴汉夔, 吕俏莹, 阿吉艾克拜尔·艾萨. 天然产物研究与开发, 2012, 24(2): 234-238
8 YIN YongQin, SHEN ZhiBin. China Pharmaceuticals, 2012, 21(2): 19-21
尹永芹, 沈志滨. 中国药业, 2012, 21(2): 19-21
9 ZOU Sheng, XU Yi, ZHANG Qing. Nat. Prod. Res. Dev., 2015, 27(8): 1501-1509
邹 胜, 徐 溢, 张 庆. 天然产物研究与开发, 2015, 27(8): 1501-1509
10 AbuReidah I M, AliShtayeh M S, Jamous R M, ArráezRomán D, SeguraCarretero A. Food Chem., 2015, 166(166): 179-191
11 Breemen R B V, Tao Y, Li W. Fitoterapia, 2011, 82(1): 38-43
12 Li D Q, Zhao J, Xie J, Li S P. J. Pharmaceut. Biomed. Anal., 2014, 88: 130-135
13 Tolonen A, Turpeinen M. Drug Discovery Today, 2009, 14(3-4): 120-133
14 Zhao H, Zhou S, Zhang M, Feng J, Wang S, Wang D, Geng Y, Wang X. J. Pharmaceut. Biomed. Anal., 2015, 120: 235-240
15 Yu F, Kong L, Zou H, Lei X. Combinatorial Chem. High Throughput Screen., 2010, 13(10): 855-868
16 Deng S, Xia , Xiao H. Chem. Commun., 2014, 50(20): 2582-2584
17 Yang X X, Xu F, Wang D, Yang Z W, Tan H R, Shang M Y, Wang X, Cai S Q. J. Chromatogr. A, 2015, 1413: 33-46
18 Zhang A, Ye F, Lu J, Zhao S. Food Chem., 2013, 141(3): 1854-1859
19 Guo L P, Jiang T F, Lv Z H, Wang Y H. J. Pharmaceut. Biomed. Anal., 2010, 53(5): 1250-1253
20 Yang J B, Tian J Y, Dai Z, Ye F, Ma S C, Wang A G. Fitoterapia, 2016, 117: 65-70
21 Ting L I, Zhang X D, Song Y W. Chin. J. Clin. Pharmacol. Therapeut., 2005, 23: 56-61
22 Zhou X, Liang J, Zhang Y, Zhao H, Guo Y, Shi S. J. Chromatogr. B, 2015, 985: 149-154
23 HE ZhongMei, WANG XiaoHui, LI GuoFeng, SUN JiaMing, YANG He, GAO YuGang, ZHANG LianXue. Chin. J. Anal. Chem., 2013, 41(11): 1694-1698
何忠梅, 王晓慧, 李国峰, 孙佳明, 杨 鹤, 郜玉钢, 张连学. 分析化学, 2013, 41(11): 1694-1698
24 Willems J L, Khamis M M, Mohammed S W, Purves R W, Katselis G, Low N H, ElAneed A. Anal. Chim. Acta, 2016, 933: 164-174
25 Luo L, Hao Y, Jia L, Zhu W. Trop. J. Pharmaceut. Res., 2016, 15(2): 405-409
26 ZHU ChunSheng, LIN ZhiJian, ZHANG Bing, NIU HongJuan, WANG XueJie, ZHANG XiaoMeng. Journal of Beijing University of Traditional Chinese Medicine, 2016, 39(3): 247-251
朱春勝, 林志健, 张 冰, 牛红娟, 王雪洁, 张晓朦. 北京中医药大学学报, 2016, 39(3): 247-251
27 YAN Hua, QIU Chen, ZHONG Kai, HUANG YiNa, GAO Hong. Modern Food Science & Technology, 2015, 31(7): 44-49
颜 欢, 邱 琛, 钟 凯, 黄毅娜, 高 鸿. 现代食品科技, 2015, 31(7): 44-49
28 Jian C, Mangelinckx S, Li M, Wang Z, Li W, Kimpe N D. Fitoterapia, 2014, 99: 1-6
29 LIU XueHui, LI MiLu, TAN Bin, CHEN HuiHeng, LU Ying. Modern Food Science and Technology, 2014, 30(3): 103-107
刘雪辉, 李觅路, 谭 斌, 陈惠衡, 陆 英. 现代食品科技, 2014, 30(3): 103-107
30 PANG MeiRong, LIU LingYi, GAO WangLei, ZHANG Ying. Chinese Traditional and Herbal Drugs, 2015, 46(2): 305-312
庞美蓉, 刘零怡, 高汪磊, 张 英. 中草药, 2015, 46(2): 305-312
31 Oboh G, Agunloye O M, Adefegha S A, Akinyemi A J, Ademiluyi A O. J. Basic Clin. Physiol. Pharmacol., 2015, 26(2): 165-170
Abstract Highaffinity αglucosidase inhibitors were screened from Cichorium glundulosum Boiss.et Hout seed (CGS) extract by ultrafiltration affinityliquid chromatographymass spectrometry (UFLCMS) and molecular docking. By taking 4nitrobenzeneαDglucopyranoside (PNPG) as substrate and acarbose as positive control to evaluate the inhibitory activity of CGS extract, IC50 of acarbose and CGS extract were 0.003 mg/mL and 0.447 mg/mL, respectively. Meanwhile, 4 compounds from CGS extract by UFLCMS were screened and identified. Then by using autodock software, the compounds that combined with αglucosidase were well screened out, including chlorogenic acid and isochlorogenic acid A. The inhibitory activity of chlorogenic acid and chlorogenic acid A against αglucosidase was verified in vitro. The results showed that the inhibitory activity of the compounds toward αglucosidase presented the sequence of acarbose>isochlorogenic acid A>chlorogenic acid. The inhibition rate of isochlorogenic acid A was close to acarbose. The experimental results illustrated that UFLCMS and molecular docking could be used to screen high affinity enzyme inhibitors from CGS.
Keywords Ultrafiltration affinity; Liquid chromatographymass spectrometry; Molecular docking; Cichorium glundulosum Boiss.et Hout seed; αGlucosidase inhibitors