
新疆一枝蒿有效部位主要成分的制备与鉴定Δ
2017-07-03 14:57戎晓娟顾政一贺金华新疆维吾尔自治区药物研究所药物分析室乌鲁木齐830004新疆医科大学药学院药剂物化教研室乌鲁木齐8300
中国药房 2017年16期
戎晓娟,顾政一,贺金华#,杨 璐(.新疆维吾尔自治区药物研究所药物分析室,乌鲁木齐 830004;.新疆医科大学药学院药剂物化教研室,乌鲁木齐 8300)
新疆一枝蒿有效部位主要成分的制备与鉴定Δ
戎晓娟1*,顾政一1,贺金华1#,杨 璐2(1.新疆维吾尔自治区药物研究所药物分析室,乌鲁木齐 830004;2.新疆医科大学药学院药剂物化教研室,乌鲁木齐 830011)
目的:建立新疆一枝蒿有效部位主要成分的快速识别和高效制备的方法,为民族药的研究提供参考。方法:采用液相-高分辨质谱(LC-HRMS/MS)法对一枝蒿有效部位的主要成分进行初步推测,利用高效液相色谱、紫外光谱和质谱法将部分化合物与相应对照品进行比对分析确定其名称;对尚未确定的化合物采用柱分离及制备液相色谱技术快速获得单体,并确定其结构。结果:从一枝蒿有效部位分离出5个化合物,鉴定出其中2个化合物为艾黄素和紫花牡荆素;获得1个单体化合物(得率为0.35mg/g,纯度为98.5%),经结构确证为6-去甲氧基-4′-O-甲基茵陈色原酮-7-O-β-D-葡萄糖苷。结论:本试验建立的方法实现了目标成分的快速分离、鉴定、制备,可应用于民族药复杂的物质基础研究。
一枝蒿;液质联用;制备液相色谱;分离;鉴定;单体制备;结构确证
中药、民族药物质基础研究是阐明其药效物质、药理作用及其机制和临床疗效的先决条件,也是深层次改进工艺和剂型、制定质量标准、提高临床疗效的重要基础,是中药现代化的重要组成部分。长期以来,对中药、民族药物质基础的研究已经取得了很大成就,但同时此项工作难度大、用时长、耗试剂,大部分时间都在分离已知化合物,做了很多重复性工作,造成大量时间和资金的浪费[1-2]。因此,能否快速识别已知成分,有目标地分离未知成分或目标化合物,从而提高工作效率,是中药、民族药研究亟待解决的问题。
一枝蒿(Artemisia rupestris L.)为菊科蒿属(Artemisia L.)植物岩蒿的地上部分或全草,为新疆道地药材,也是维吾尔医常用传统药材,在我国主要分布在新疆北疆一带[3]。一枝蒿具有清热解毒、健胃消食、镇静止吐、抗过敏等功效[4]。本课题组前期对新疆一枝蒿黄酮类提取物进行了抗乙肝病毒试验研究并确定了其有效部位[5-6],在本试验中,则对该有效部位的主要成分进行研究,以明确其成分组成并获得一定量的单体,为后续研究提供物质基础。
将液质联用技术和柱色谱、制备液相色谱技术结合起来研究中药、民族药的物质基础,是本课题组经过反复试验和探索后总结的研究思路,即首先利用具有高效分离分析能力的液相-高分辨质谱(LC-HRMS/MS)法对一枝蒿有效部位的成分进行初步分析,推测可能化合物;并利用高效液相色谱(HPLC)、紫外光谱(UV)和质谱法(MS)与已有的对照品进行比对,确定部分化合物的结构;继而将传统柱分离技术与具有高分离能力的制备液相色谱结合,快速、高效地获得一定量纯度较高的单体化合物,并对其进行结构确认。
1 材料
1.1 仪器
BP211D电子天平(德国赛多利斯公司);Dionex UltiMate 3000 HPLC系统、二极管阵列检测器(DAD)、四级杆-静电场轨道阱质谱仪(Q-Exactivemass spectrometer)配备Xcalibur 2.3软件(美国Thermo公司);2545二元梯度系统制备型液相色谱仪(美国Waters公司);INOVA-500核磁共振波谱仪(美国Varian公司);SK3300H超声波清洗仪(上海科导超声仪器有限公司)。
1.2 药材、对照品与试剂
一枝蒿(新疆西部加斯特药业有限公司,批号:20130823,由新疆维吾尔自治区药物研究所民族药室何江副研究员鉴定为真品);艾黄素对照品(批号:111879-201001,纯度:98.3%)、紫花牡荆素对照品(批号:111871-201102,纯度:99.1%)来源于中国食品药品检定研究院;甲醇、甲酸为色谱纯,水为超纯水,其余试剂均为分析纯。
2 方法与结果
2.1 一枝蒿黄酮类抗乙肝病毒有效部位的制备
将一枝蒿药材粉碎,加入10倍量50%乙醇,加热回流提取3次,提取时间分别为2、2、1 h,合并提取液,滤过,减压浓缩,真空干燥,得浸膏。将浸膏粉末用水混悬,上样于聚酰胺柱,依次用水和10%、30%、50%、70%、95%乙醇进行洗脱,收集各洗脱液,将50%乙醇洗脱液减压浓缩、干燥,得50%乙醇洗脱物,即为有效部位[5]。
2.2 采用LC-HRMS/MS法对有效部位进行成分分析
2.2.1 供试品溶液的制备 称取有效部位粉末适量,加入50%甲醇水适量,超声30m in(功率:160W、频率:59 kHz)至完全溶解,冷却至室温,定容,制成约2mg/m L的溶液,过微孔滤膜(0.2µm),取续滤液,即得。
2.2.2 色谱条件 色谱柱:Agilent C18(250 mm×4.6 mm,5µm);流动相:水(A)-甲醇(B),梯度洗脱(0~40 m in,20%B→85%B;40~45 m in,85%B;45~55 m in,20%B);检测波长:200~400 nm;流速:1.0m L/m in;进样量:10μL;柱温:30。柱后按1∶2分流,其中1/3进入MS仪,2/3进入废液瓶。
2.2.3 MS条件 采用正、负离子全扫描模式,扫描范围为质荷比(m/z)100~1 500。电喷雾离子源雾化温度为300,毛细管电压为3 500~3 300V,离子传输管温度为220,鞘气压力为40 arb,辅助气压力为11 arb,分辨率(R)为35 000。
2.2.4 LC-HRMS/MS法分析结果推测 对有效部位进行分析后,出现5个化合物峰,结果见图1、图2;根据文献[7-11]报道的一枝蒿化学成分推测出5个化合物的可能名称,结果见表1。
2.3 2号、4号化合物与对照品比对
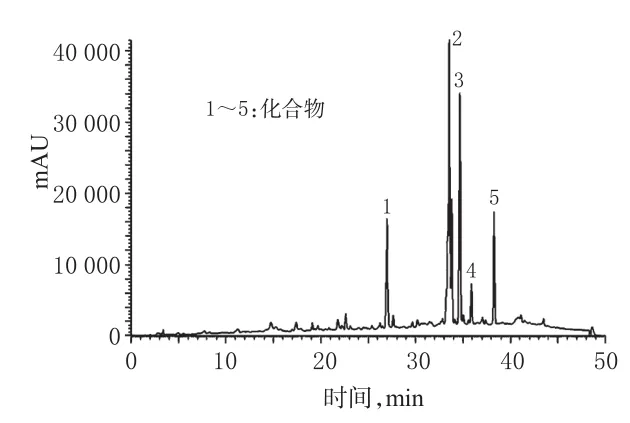
图1 供试品HPLC图Fig 1 HPLC chromatogramsof testsam ples

图2 供试品总离子流色谱图(正离子检测模式)Fig 2 Total ion flow chromatogram s of test sam p les(positive ion detectionmode)
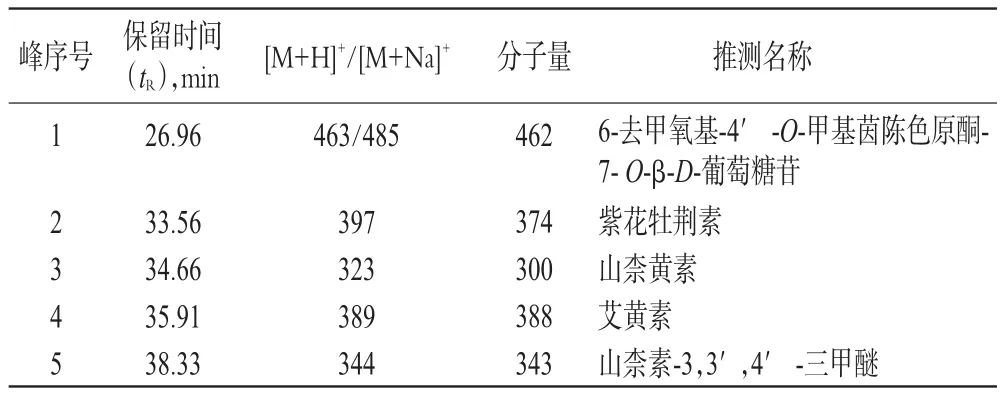
表1 5个化合物推测结果Tab 1 Speculative resultof5 compounds
根据上述推测结果,对各化合物进行进一步的比对和确认。由于目前只有紫花牡荆素和艾黄素有市售对照品,故首先对2号和4号化合物进行分析。
2.3.1 2号化合物与紫花牡荆素对照品比对 (1)溶液的制备。精密称取紫花牡荆素对照品约5mg,置于10 m L量瓶中,加甲醇溶解并定容,制成质量浓度为0.5 mg/m L的溶液,即得紫花牡荆素对照品。供试品溶液制备同“2.2.1”项下。(2)色谱条件。色谱柱:Agilent C18(250mm×4.6mm,5µm);流动相:0.1%甲酸水(A)-乙腈(B),梯度洗脱(0~40 m in,10%B→90%B;40~45 min,90%B;45~55m in,10%B);检测波长:330 nm;流速:1.0m L/min;进样量:10μL;柱温:30。(3)HPLC和UV图谱比对。在上述“(2)”项色谱条件下,取紫花牡荆素对照品与供试品溶液进样分析,结果2号峰与紫花牡荆素对照品保留时间(分别为25.263、25.265m in)及UV吸收曲线一致,证实2号化合物即为紫花牡荆素。图谱详见图3、图4。
2.3.2 4号化合物与艾黄素对照品比对 (1)溶液的制备。精密称取艾黄素对照品约3mg,置于10m L量瓶中,加甲醇溶解并定容,制成质量浓度为0.3mg/m L的溶液,即得艾黄素对照品溶液。供试品溶液制备同“2.2.1”项下。(2)色谱条件。同“2.3.1(2)”项下。(3)HPLC和UV图谱比对。在“2.3.1(2)”项色谱条件下,取艾黄素对照品与供试品溶液进样分析,结果4号峰与艾黄素对照品保留时间(分别为28.633、28.638min)及UV吸收曲线一致,证实4号化合物即为艾黄素。图谱详见图5、图6。

图3 2号化合物与紫花牡荆素对照品HPLC比对图Fig 3 HPLC chromatogram com parison of com pound 2 and casticin reference substance
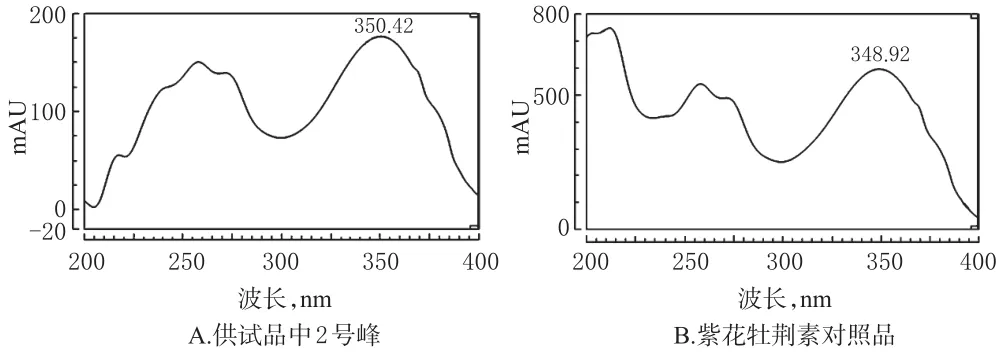
图4 2号化合物与紫花牡荆素对照品UV比对图Fig 4 UV chrom atogram com parison of com pound 2 and casticin reference substance

图5 4号化合物与艾黄素对照品HPLC比对图Fig 5 HPLC chromatogram com parison of com pound 4 and artem etin reference substance
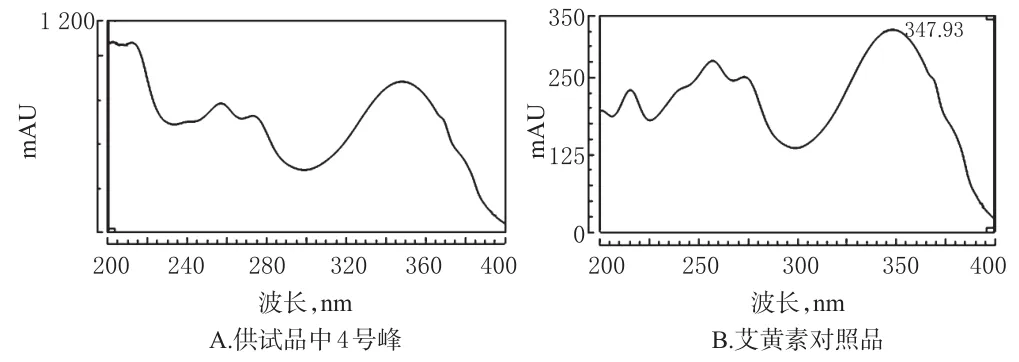
图6 4号化合物与艾黄素对照品UV比对图Fig 6 UV chromatogram comparison of com pound 4 and artemetin reference substance
2.4 单体制备及鉴定
上文利用HPLC、UV、MS法将可能化合物与相应对照品进行比对分析,确定了2号和4号化合物;但1号、3号和5号化合物名称仍然未知,需要通过柱分离及制备液相色谱获得单体,并确定结构。
2.4.1 柱分离得粗分物 将有效部位制成粉末,用水混悬,依次用石油醚和三氯甲烷等体积萃取,将三氯甲烷萃取液合并,经旋转蒸发仪浓缩,得浸膏备用;取一枝蒿浸膏与硅胶以1∶1.5质量比进行吸附并挥干,湿法上样于硅胶柱(径高比为1∶8),依次用10∶0、9∶1、8∶2、7∶3、6∶4、5∶5、4∶6、3∶7、2∶8、1∶9、0∶10比例的石油醚-乙酸乙酯洗脱,洗脱流速为1倍柱体积(BV)/h。分段收集,薄层色谱法监测成分情况,在7∶3部分的洗脱液中有明显清晰斑点,故将此部分收集、浓缩,经重结晶纯化后获得粗分物。经HPLC分析,粗分物的保留时间与供试品中的1号峰保留时间一致,故确定为1号化合物,色谱纯度为88.0%,达不到采用核磁进行结构鉴定的纯度要求,需用制备液相色谱进一步纯化。
2.4.2 制备液相色谱分离纯化得单体化合物 为获得高纯度的单体,将1号粗分物进一步采用高压制备液相技术进行分离纯化。(1)样品的制备。称取1号粗分物粉末于量瓶中,加入70%甲醇,超声20min(功率:160W,频率:59 kHz)使溶解,制成约1mg/m L的溶液,过滤,即得。(2)制备液相色谱条件。制备型色谱柱:Xbridge Prep C1(8250mm×19mm,5µm);流动相:甲醇(A)-水(B),梯度洗脱(0~11m in,78%A;11~12m in,95%A;12~14min,78%A);流速:9m L/min;进样量:2.0m L;检测波长:347 nm。(3)HPLC分析条件。色谱柱:Phenomenex Luna C1(8250mm×4.6mm,5µm);流动相:乙腈(A)-0.1%甲酸水溶液(B),梯度洗脱(0~40 min,10%A→90%A;40~45min,90%A;45~47min,10% A);流速:1m L/m in;检测波长:347 nm;进样量:10μL;柱温:30。结果,经上述制备条件,将1号粗分物制备多份,反复进样,收集样品;获得的样品经上述分析条件测定,色谱纯度为98.5%,按一枝蒿药材计得率为0.35 mg/g。
2.4.3 1号化合物结构确认 经MS和核磁共振波谱进行分析,解析1号化合物的结构。
1号化合物:黄色无定形粉末,电子电离质谱(EIMS)m/z:462,与分子式C22H22O11相符。氢谱(1H-NMR)[二甲基亚砜(DMSO),400 MHz]化学位移(δ):5.10(1H,s,H-3),6.46(1H,d,J=2,H-6),6.72(1H,s,H-8),7.36(1H,d,J=4,H-2′),7.09(1H,d,J=4,H-3′),7.09(1H,d,J=4,H-5)′,7.36(1H,d,J=4,H-6′),3.80(3H,s,—OCH3),12.81(1H,s,—OH)。13C-NMR(DMSO,125 MHz)δ:168.32(C-2),87.60(C-3),183.37(C-4),161.22(C-5),100.24(C-6),162.89(C-7),94.95(C-8),154.70(C-9),103.85(C-10),144.46(C-1)′,122.12(C-2)′,115.60(C-3)′,158.00(C-4)′,115.60(C-5)′,122.12(C-6)′,100.02(Glc-1),73.25(Glc-2),76.57(Glc-3),69.75(Glc-4),77.33(Glc-5),60.80(Glc-6),55.78(C-4′,—OCH3)。结合文献[12],确定该化合物为6-去甲氧基-4′-O-甲基茵陈色原酮-7-O-β-D-葡萄糖苷,化学结构式见图7。
3 讨论
本试验研究所用的新疆一枝蒿黄酮类提取物经药效试验证实其具有较强的抗乙肝病毒活性[5],故对其物质基础进行深入研究意义较大。在分离目标化合物时,首先选择传统的柱色谱进行粗分,再选择制备液相色谱进行纯化,将前者分离上样量大与后者分离能力强相结合,能快速、高效地获得纯度较高的单体,更适合研究复杂的中药、民族药的物质组成。

图7 6-去甲氧基-4′-O-甲基茵陈色原酮-7-O-β-D-葡萄糖苷结构式Fig 7 Chem ical structural form ula of 6-demethoxy-4′-O-m ethoxy-capillarisin-7-O-β-D-glucoside
另外,由于时间限制本试验尚未对3号和5号化合物进行分离制备和结构确认,后续将借鉴本文方法和思路进行该项工作。
综上,本试验采用的研究思路目标明确、高效快速,不仅可以实现目标成分的快速分离鉴定,而且可以实现活性组分的快速制备,获得的较高纯度的单体成分将为一枝蒿作用机制的研究提供物质保障。
[1] 崔凤侠,章宸,姜勇,等.新疆一枝蒿乙酸乙酯部位化学成分研究[J].中国中药杂志,2013,38(11):1757-1759.
[2] 屠鹏飞,史社坡,姜勇.中药物质基础研究思路与方法[J].中草药,2012,43(2):209-215.
[3] 国家中医药管理局《中华本草》编委会.中华本草:维吾尔药卷[M].上海:上海科学技术出版社,2005:47-48.
[4] 国家药典委员会.中华人民共和国卫生部药品标准:中药材:第一册[S].北京:人民卫生出版社,1992:1.
[5] 杨璐,张素挽,贺金华,等.一枝蒿黄酮类提取物体内抗乙肝病毒活性研究及化学成分分析[J].新疆医科大学学报,2016,39(5):578-581.
[6] 秦子茹,贺金华,顾政一,等.一枝蒿不同溶剂提取物抗病毒作用的谱效关系研究[J].中国药房,2015,26(7):889-893.
[7] 吉腾飞,杨建波,宋卫霞,等.新疆一枝蒿化学成分研究Ⅱ[J].中国中药杂志,2007,32(12):1184-1189.
[8] 宋卫霞,吉腾飞,司伊康,等.新疆一枝蒿化学成分的研究[J].中国中药杂志,2006,31(21):1790-1792.
[9] 杨建波,吉腾飞,宋卫霞,等.新疆一枝蒿化学成分的研究[J].中草药,2008,39(8):1125-1127.
[10] 杨建波,苏亚伦,吉腾飞,等.HPLC法测定新疆一枝蒿全草中一枝蒿酮酸的含量[J].药物分析杂志,2010,30(1):103-105.
[11] 汪豪,杜慧斌,朱峰妍,等.新疆一枝蒿化学成分的研究[J].中国药科大学学报,2011,42(4):310-313.
[12] 王燕,吐尔洪·阿西木,堵年生.新疆一枝蒿化学成分的研究[J].新疆医科大学学报,2004,27(4):361-363.
Preparation and Identification of M ain Ingredients in Effective Parts of Xinjiang Artem isia rupestris
RONG Xiaojuan1,GU Zhengyi1,HE Jinhua1,YANG Lu2(1.Dept.of Pharmaceutical Analysis,the Xinjiang Institute of Material Medica,Urumqi 830004,China;2.Dept.of Pharmacology and Physiology,College of Pharmacy,Xinjiang Medical University,Urumqi830011,China)
Artemisia rupestris;LC-MS;Preparative liquid chromatography;Separation;Identification;Monomer preparation;Structure identification
R284.2
A
1001-0408(2017)16-2227-04
2016-09-20
2017-02-20)
(编辑:刘 萍)
国家自然科学基金新疆联合基金资助项目(No. U1303224)
*助理研究员,硕士。研究方向:药物分析。电话:0991-2326572。E-mail:109303620@qq.com
#通信作者:研究员,硕士。研究方向:药物分析。电话:0991-2326572。E-mail:hejh1216@163.com
DOI10.6039/j.issn.1001-0408.2017.16.18
ABSTRACTOBJECTIVE:To establish amethod for rapid identification and efficient preparation ofmain ingredients in effective parts of Xinjiang Artemisia rupestris,and provide reference for researching the ethnic medicines.METHODS:LC-HRMS/MSwas conducted for the preliminary study ofmain ingredients in effective parts of A.rupestris.HPLC,UV and MSwere used to compare and analyze parts of the compounds and its reference substances,their names were determ ined.Column separation and preparative liquid chromatography were used for the undeterm ined compounds to receive monomer rapidly,and the structures were identified. RESULTS:5 compounds were separated from the effective parts,2 of which were identified as artemetin and casticin.A monomeric compound was obtained(yield was 0.35mg/g,the purity was 98.5%),which was confirmed to be 6-demethoxy-4′-O-methoxycapillarisin-7-O-β-D-glucoside by the structure.CONCLUSIONS:The method has achieved rapid separation,identification and preparation of target ingredients,which can be used for the fundamental research of ethnicmedicine complex materials.
猜你喜欢
山西医科大学学报(2021年10期)2021-11-18
天津大学学报(自然科学与工程技术版)(2021年9期)2021-06-01
广西医科大学学报(2021年4期)2021-05-25
扬子江诗刊(2020年3期)2020-11-17
农产品加工(2020年17期)2020-10-22
农民致富之友(2020年16期)2020-06-19
扬子江(2020年3期)2020-06-08
世界科学技术-中医药现代化(2020年10期)2020-04-06
植物研究(2020年1期)2020-03-10
世界中医药(2019年2期)2019-09-10
