
普乐士泰胶囊的质量标准研究Δ
2017-07-07 15:09叶绿萍龚敏阳唐春丽马儒清广西中医药大学第一附属医院南宁530023
中国药房 2017年15期
叶绿萍,宾 彬,龚敏阳,唐春丽,马儒清(广西中医药大学第一附属医院,南宁 530023)
普乐士泰胶囊的质量标准研究Δ
叶绿萍*,宾 彬,龚敏阳,唐春丽,马儒清(广西中医药大学第一附属医院,南宁 530023)
目的:建立普乐士泰胶囊的质量标准。方法:采用薄层色谱法(TLC)对制剂中太子参、石菖蒲、大血藤、茜草、王不留行进行定性鉴别;采用高效液相色谱法测定制剂中益母草碱的含量:色谱柱为AgilentEclipse XDB-C18,流动相为甲醇-0.025mol/L磷酸二氢钾溶液(用磷酸调pH至2.5)(24∶76,V/V),流速为1.0m L/min,检测波长为277 nm,柱温为30℃,进样量为10μL。结果:太子参、石菖蒲、大血藤、茜草、王不留行的TLC图斑点清晰,分离度好,阴性对照无干扰。盐酸益母草碱检测质量浓度线性范围为4.05~81.00μg/m L(r=0.999 9);精密度、稳定性、重复性试验的RSD<2.0%;加样回收率为98.47%~103.83%(RSD=2.04%,n=9)。结论:该研究所建标准可用于普乐士泰胶囊的质量控制。
普乐士泰胶囊;质量标准;薄层色谱法;益母草碱;高效液相色谱法
普乐士泰胶囊是由广西中医药大学第一附属医院宾彬教授根据临床经验方研制而成。其处方以益母草为君药,川牛膝、薏苡仁、苦杏仁为臣药,太子参、石菖蒲、白蔻仁、大血藤、败酱草等多味中药相佐使,具有活血化瘀、清热通淋、化湿导浊、行气止痛的功效,多年来在临床上用于治疗慢性前列腺炎具有明显的疗效[1-2],为医院制剂。笔者经反复试验及参阅文献,采用薄层色谱法(TLC)对普乐士泰胶囊中太子参、石菖蒲、大血藤、茜草、王不留行进行定性鉴别,并采用高效液相色谱法(HPLC)对制剂中益母草碱含量进行测定[3],以有效控制该制剂的质量。
1 材料
1.1 仪器
1200型HPLC仪,包括二极管阵列检测仪(美国Agilent公司);GR-202型电子分析天平(日本AND公司);Simplicity TM型超纯水系统(美国M illipore公司);KQ-100DV型数控超声波清洗器(昆山市超声仪器有限公司,功率:250W,频率:40 kHz)。
1.2 药品与试剂
普乐士泰胶囊(广西中医药大学第一附属医院制剂室 自 制 ,批 号 :20140107、20140116、20140117、20140121、20140123,规格:0.38 g/粒);盐酸益母草碱对照品(中国食品药品检定研究院,批号:111823-201202,纯度:94.7%);太子参、石菖蒲、大血藤、茜草、王不留行对照药材(广西万宝堂药业有限公司,批号分别为121014-201408、121098-201406、121353-201401、121049-201404、121094-201405);硅胶G薄层板(青岛海洋化工有限公司);甲醇为色谱纯,乙醇、磷酸、磷酸二氢钾均为分析纯,水为超纯水。
2 方法与结果
2.1 定性鉴别
2.1.1 太子参 取10粒样品内容物,加甲醇10m L,超声温浸(42℃)提取30m in,滤过并浓缩至1m L,作为供试品溶液。另取太子参对照药材粉末(过2号筛)1 g,同供试品溶液制备方法制备对照药材溶液。按普乐士泰胶囊处方和工艺制备缺太子参的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][4]试验,吸取上述3种溶液各4 μL,分别点于同一硅胶G薄层板上,以正丁醇-冰醋酸-水(4∶1∶1,V/V/V)为展开剂,预饱和15m in,展开,取出,晾干,喷以0.2%茚三酮试液,置于恒温箱中,以105℃加热至斑点显色清晰,置日光灯下检视。结果,供试品色谱中,在与对照药材色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图1A。
2.1.2 石菖蒲 取8粒样品内容物,置于圆底烧瓶中,加入石油醚(60~90℃)30m L,水浴加热回流1 h,滤过,滤液水浴蒸干,加石油醚(60~90℃)1m L溶解药渣,作为供试品溶液。另取石菖蒲对照药材粉末(过2号筛)0.2 g,同供试品溶液制备方法制备对照药材溶液。按普乐士泰胶囊处方和工艺制备缺石菖蒲的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][4]试验,吸取上述3种溶液各3μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-石油醚(60~90℃)(1∶4,V/V)为展开剂,预饱和15min,展开,取出,晾干,常温下放置1 h,以碘蒸气熏至斑点显色清晰,置日光灯下检视。结果,供试品色谱中,在与对照药材色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图1B。

图1 薄层色谱图Fig 1 TLC chromatograms
2.1.3 大血藤 取20粒样品内容物,加甲醇25m L,超声温浸(42℃)提取30min,滤过,滤液水浴蒸干,加2%氢氧化钠溶液5m L溶解药渣,以盐酸调pH至2.0,再加乙醚5m L充分振摇提取,用分液漏斗分离,共3次,合并乙醚液,挥干,加甲醇1 m L溶解药渣,作为供试品溶液。另取大血藤对照药材粉末(过2号筛)2.5 g,同供试品溶液制备方法制备对照药材溶液。按普乐士泰胶囊处方和工艺制备缺大血藤的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][4]试验,吸取上述3种溶液各4μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲酸-丙酮(8∶1∶1,V/V/V)为展开剂,预饱和15m in,展开,取出,晾干,喷以2%三氯化铁乙醇溶液,置紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图1C。
2.1.4 茜草 取10粒样品内容物,加甲醇15m L,超声温浸(42℃)提取30m in,滤过,滤液水浴蒸干,加甲醇1 m L溶解药渣,作为供试品溶液。另取茜草对照药材粉末(过2号筛)2.5 g,同供试品溶液制备方法制备对照药材溶液。按普乐士泰胶囊处方和工艺制备缺茜草的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][4]试验,吸取上述3种溶液各5μL,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-丙酮(4∶1,V/V)为展开剂,预饱和15m in,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图1D。
2.1.5 王不留行 取12粒样品内容物,加甲醇25m L,水浴回流提取30m in,放冷,滤过,滤液水浴蒸干,加1 m L甲醇溶解药渣,作为供试品溶液。另取王不留行对照药材粉末(过2号筛)2.5 g,同供试品溶液制备方法制备对照药材溶液。按普乐士泰胶囊处方和工艺制备缺王不留行的阴性样品,并按供试品溶液制备方法制成阴性对照溶液。按TLC法[2015年版《中国药典》(四部)][4]试验,吸取上述3种溶液各4μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(15∶7∶2,V/V/V)充分振摇静置的下层液为展开剂,预饱和15m in,展开,取出,晾干,喷以碘化铋钾试液,置日光灯下检视。结果,供试品色谱中,在与对照药材色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图1E。
2.2 含量测定
2.2.1 色谱条件与系统适用性试验 色谱柱:Agilent Eclipse XDB-C18(250mm×4.6mm,5μm);流动相:甲醇-0.025mol/L磷酸二氢钾溶液(用磷酸调pH至2.5)(24∶76,V/V);流速:1.0m L/min;检测波长:277 nm;柱温:30℃;进样量:10μL[5-6]。在上述色谱条件下,理论板数以益母草碱(以盐酸益母草碱计)峰计不少于5 000;各成分基线分离良好,分离度>1.5,详见图2。
2.2.2 对照品溶液的制备 取对照品适量,精密称定,加流动相制成盐酸益母草碱质量浓度为81.00μg/m L的对照品溶液。
2.2.3 供试品溶液的制备 取10粒样品内容物,研匀,取约2粒的量,精密称定,精密加50%乙醇溶液25m L,称定质量,超声温浸(42℃)处理60m in,放冷,加50%乙醇溶液补足减失的质量,滤过,精密量取续滤液10m L,水浴蒸干,残渣加流动相溶解,移至10m L量瓶中,加流动相定容,经0.45μm微孔滤膜滤过,即得。
2.2.4 阴性对照溶液的制备 按普乐士泰胶囊处方和工艺制备缺益母草的阴性样品,并按“2.2.3”项下方法制成阴性对照溶液。

图2 高效液相色谱图Fig 2 HPLC chromatograms
2.2.5 线性关系考察 精密量取“2.2.2”项下对照品溶液各适量,分别置于20m L量瓶中,加流动相定容,制成盐酸益母草碱质量浓度分别为4.05、8.10、16.20、40.50、60.75、81.00μg/m L的系列对照品溶液。精密量取上述系列对照品溶液各10μL,按“2.2.1”项下色谱条件进样测定,记录峰面积。以盐酸益母草碱质量浓度(x,μg/m L)为横坐标、峰面积(y)为纵坐标进行线性回归,得回归方程y=0.055x+0.137(r=0.999 9)。结果表明,盐酸益母草碱检测质量浓度线性范围为4.05~81.00 μg/m L。
2.2.6 精密度试验 取“2.2.2”项下对照品溶液适量,按“2.2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,盐酸益母草碱峰面积的RSD=1.10%(n=6),表明仪器精密度良好。
2.2.7 稳定性试验 取“2.2.3”项下供试品溶液(批号:20140107)适量,分别于室温下放置0、2、4、8、12、24 h时按“2.2.1”项下色谱条件进样测定,记录峰面积。结果,盐酸益母草碱峰面积的RSD=0.93%(n=6),表明供试品溶液室温下放置24 h内基本稳定。
2.2.8 重复性试验 精密称取同一批样品(批号:20140107)适量,按“2.2.3”项下方法制备供试品溶液,共6份,再按“2.2.1”项下色谱条件进样测定,记录峰面积并计算含量。结果,盐酸益母草碱平均含量为0.087 1mg/m L,RSD=1.27%(n=6),表明本方法重复性良好。
2.2.9 加样回收率试验 取已知含量样品(批号:20140107)适量,共9份,分别加入低、中、高质量的盐酸益母草碱对照品各适量,按“2.2.3”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表1。
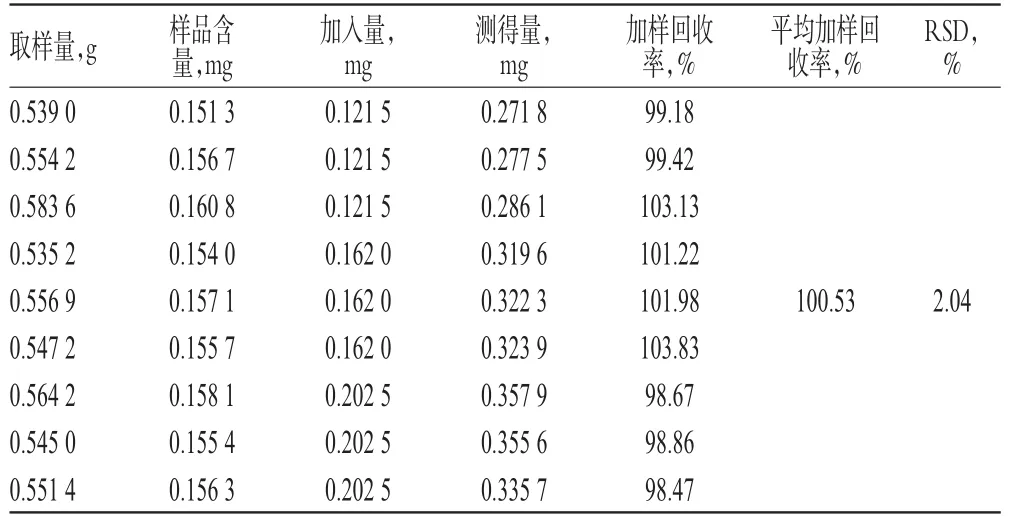
表1 加样回收率试验结果(n=9)Tab 1 Resultsof recovery tests(n=9)
2.2.10 样品含量测定 取6批样品各适量,分别按“2.2.3”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并计算样品含量,结果见表2。

表2 样品含量测定结果(n=6,m g/粒)Tab 2 Results of content determination of samples(n=6,mg/capsule)
3 讨论
3.1 TLC条件
TLC法是利用吸附剂对样品中各成分吸附能力不同,及展开剂对其解吸附能力的不同,使各成分达到分离目的[7]。普乐士泰胶囊中含有多味药材,成分复杂,影响各成分分离干扰因素众多。根据不同药味及其化学成分的物理特性[8],笔者对样品的前处理方法及展开剂的组方配比进行了反复研究,并采用自制硅胶G板和商品板反复考察了其重复性,最终确定了本研究中各药味的TLC鉴别方法,其能达到较好的定性鉴别效果。
3.2 定量指标成分的选择
普乐士泰胶囊中益母草为君药,除具有活血化瘀、调经利水的功效外,还有异于传统适应证的溶栓、抗凝、降脂、扩张冠脉、降血黏度、改善微循环、抗炎镇痛、利尿和增强小肠平滑肌收缩等诸多作用[9]。这些药理作用与其所含生物碱密切相关,代表性的活性成分如盐酸水苏碱(Stachydrine)和益母草碱(Leonurine)。2015年版《中国药典》(一部)[8]将此两成分同时作为益母草的含量测定指标成分,分别制定了相应的HPLC测定方法,并规定了最低含量标准。益母草碱是存在于益母草中的特异生物碱,与其他生物碱相比更具专属性[10]。但不同品种和采收季节的益母草中益母草碱含量相当不均衡,故选择益母草碱作为普乐士泰胶囊的质控指标更有意义。
3.3 流动相的选择
选择流动相时曾经用不同配比的甲醇-水和不同的流速来考察,结果色谱峰有拖尾现象,且待测成分的理论板数低,后改用甲醇-磷酸二氢钾溶液(24∶76,V/V),但是待测成分色谱峰却出现了双峰现象,结果不理想。故笔者通过采用调节流动相pH的方法来改良峰形并提高色谱峰的理论板数。经过反复摸索流动相的组成比例后,发现以甲醇-0.025mol/L磷酸二氢钾溶液(用磷酸调pH至2.5)(24∶76,V/V)时,出峰最佳,可基线分离,达到了样品含量测定的基本要求[11]。
3.4 检测波长的选择
[11],取盐酸益母草碱对照品适量,用本试验的流动相为溶剂制备对照品溶液,以流动相为空白在200~400 nm紫外区域内扫描,结果显示,盐酸益母草碱在277 nm波长处出现最大吸收。
综上所述,本研究所建标准可用于普乐士泰胶囊的质量控制。
参考文献
[1] 宾彬,潘章宇.普乐士泰胶囊治疗慢性非细菌性前列腺炎30例[J].河南中医,2006,26(8):42-43.
[2] 陈香玲,陈红军.高效液相色谱法同时测定八珍益母胶囊中10种活性成分含量[J].中南药学,2016,14(3):306-309.
[3] 袁承军,浩宇,俞瑜,等.高效液相色谱法测定益母草碱大鼠血浆浓度及其药动学研究[J].中国药业,2015,24(20):34-35.
[4] 国家药典委员会.中华人民共和国药典:四部[S].2015年版.北京:中国医药科技出版社,2015:57.
[5] 瞿京红,刘菁,吴进,等.高效液相色谱法测定益母草软胶囊中益母草碱的含量[J].中国医药导刊,2011,13(9):1631-1632.
[6] 李超.改进HPLC法测定吲哚美辛肠溶片中的有关物质[J].中国药房,2016,27(27):3861-3864.
[7] 高峰.TLC法和HPLC法分析黄柏配方颗粒中是否混有关黄柏[J].黑龙江医药,2016,29(1):9-11.
[8] 国家药典委员会.中华人民共和国药典:一部[S].2015年版.北京:中国医药科技出版社,2015:290-291.
[9] 贺春晖,赵懿清,李霞,等.益母草碱药理作用的分子机制研究进展[J].中国临床药理学与治疗学,2013,18(12):1419-1422.
[10] 张文婷,黄伟,王伟胜,等.不同采收期童子益母草中生物碱含量比较[J].医药导报,2013,32(2):218-220.
[11] 江帆,聂晶,余建清,等.高效液相法测定益母草中益母草碱含量研究[J].分析科学学报,2008,24(3):347-349.
Study on Quality Standard of ProstatitisCapsules
YE Lüping,BIN Bin,GONG M inyang,TANG Chunli,MA Ruqing(The First Affiliated Hospital of Guangxi University of TCM,Nanning 530023,China)
OBJECTIVE:To establish the quality standard for Prostatitis capsules.METHODS:TLC method was performed to qualitatively identify Pseudostellaria heterophylla,Acortw tatarinowii,Sargentodoxa cuneata,Rubia cordifolia and Vaccaria segetalis in the preparation.HPLC method was adopted to determine the content of leonurine in the preparation.The determ ination was performed on Agilent Eclipse XDB-C18column w ith mobile phase consisted of methanol-0.025 mol/L potassium dihydrogen phosphate(pH adjusted to 2.5)(24∶76,V/V)w ith phosphoric acidat the flow rate of 1.0 m L/min.The detection wavelength was set at 277 nm,and column temperature was 30℃.The sample size was 10μL.RESULTS:The TLC spots of P.heterophylla,A.tatarinowii,S.cuneate,R.cordifolia,and V.segetalis were clear and well-separated w ithout interference from negative control.The linear range of leonurine were 4.05-81.00μg/m L(r=0.999 9).RSDs of precision,stability and reproducibility tests were all lower than 2.0%.The recovery was 98.47%-103.83%(RSD=2.04%,n=9).CONCLUSIONS:Established standard can be used for quality control of Prostatitis capsules.
Prostatitis capsules;Quality standard;TLC;Leonurine;HPLC
R927
A
1001-0408(2017)15-2104-04
2016-11-01
2017-03-06)
(编辑:张 静)
广西壮族自治区卫生厅中医药科技专项课题(No. GZYZ1105)
*副主任药师。研究方向:临床药学、药剂学。电话:0771-5840015。E-mail:164589433@qq.com
DOI 10.6039/j.issn.1001-0408.2017.15.25
猜你喜欢
今日农业(2022年2期)2022-11-16
环球时报(2022-05-18)2022-05-18
中国药学药品知识仓库(2022年8期)2022-05-09
中国典型病例大全(2022年9期)2022-04-19
中国典型病例大全(2022年9期)2022-04-19
中草药(2022年1期)2022-01-13
今日农业(2021年6期)2021-06-09
特种经济动植物(2021年4期)2021-04-19
婚育与健康(2020年11期)2020-12-23
妇女生活(2017年10期)2017-10-10
