
高压下富氢化合物的结构与奇异超导电性∗
2017-08-01 08:48段德芳马艳斌邵子霁谢慧黄晓丽刘冰冰崔田
物理学报 2017年3期
段德芳 马艳斌 邵子霁 谢慧 黄晓丽 刘冰冰 崔田
(吉林大学物理学院,超硬材料国家重点实验室,长春 130012)
(2016年11月16日收到;2016年12月3日收到修改稿)
专题:高压下物质的新结构与新性质研究进展
高压下富氢化合物的结构与奇异超导电性∗
段德芳 马艳斌 邵子霁 谢慧 黄晓丽 刘冰冰 崔田†
(吉林大学物理学院,超硬材料国家重点实验室,长春 130012)
(2016年11月16日收到;2016年12月3日收到修改稿)
在富氢化合物中,一方面由于非氢元素的存在会对氢的子晶格产生化学预压作用,这些体系比纯氢更容易金属化.另一方面由于含氢量较多,富氢化合物可能会具有像金属氢那样较高的超导转变温度,有望成为超导家族的新成员—–氢基超导体.高压下富氢化合物的结构及超导电性已成为物理、材料等多学科的研究热点,最近理论和实验发现硫氢化合物在高压下的超导转变温度达到200 K,创造了高温超导新纪录,进一步推动了人们对富氢化合物超导电性的研究.本文主要介绍了近年来高压下几种典型富氢化合物的结构、稳定性、原子间相互作用、金属化及超导电性,希望未来能在富氢化合物中寻找到具有更高超导转变温度的超导体.
高压,富氢化合物,晶体结构,超导电性
1 引 言
1911年,荷兰物理学家Onnes首次在4.2 K的低温附近观测到了汞的超导现象,自此寻找高温超导体成为物理与材料领域的热点课题.2015年,Marzin[1]在《Nature》上发表文章,总结了继传统超导体发现以来的四个重要阶段,如图1所示.第一个阶段,1986年发现了超导转变温度(Tc)35 K的铜氧化物超导体[2],很快包括中国科学家在内的研究团队将铜氧化物超导体的Tc提升到90 K以上[3−5],高压下更是被提高到164 K[6],实现了液氮温区(77 K)超导体的梦想.赵忠贤院士等科学家凭借“液氮温区氧化物超导体的发现及研究”荣获1989年度国家自然科学一等奖.第二个阶段,2001年在简单层状结构MgB2中发现超导临界温度为39 K[7],深入研究发现其是电声相互作用驱动的传统Bardeen-Cooper-Schrieff er(BCS)理论超导体.第三个阶段,2008年发现了铁基超导体[8],随后中国科学家抓住机遇,发现一系列转变温度40 K以上的铁基超导体[9,10],并发现高压下Tc可以提高到55 K[11],尽管比铜氧化物超导体的最高临界温度低,但是改变了磁性离子(Fe离子)对超导不利的观点,为探索新的超导体开阔了思路.赵忠贤院士、陈仙辉院士、王楠林、闻海虎和方忠为代表的中国科学家凭借“40 K以上铁基高温超导体的发现及若干基本物理性质研究”荣获2013年度国家自然科学一等奖.第四个阶段,最近在高压下发现富氢化合物H3S最高超导转变温度达到203 K(−70◦C)[12,13],打破了此前铜基超导体164 K的温度纪录,与MgB2一样是传统BCS理论超导体,为获得室温超导体迈出了坚实的一步.除此之外,薛其坤院士等[14]还发现外延于钛酸锶衬底上的单层FeSe薄膜,由于界面增强效应,其超导转变温度可以达到70 K,远高于体材料,被称为界面高温超导体,开拓了高温超导领域的新方向.
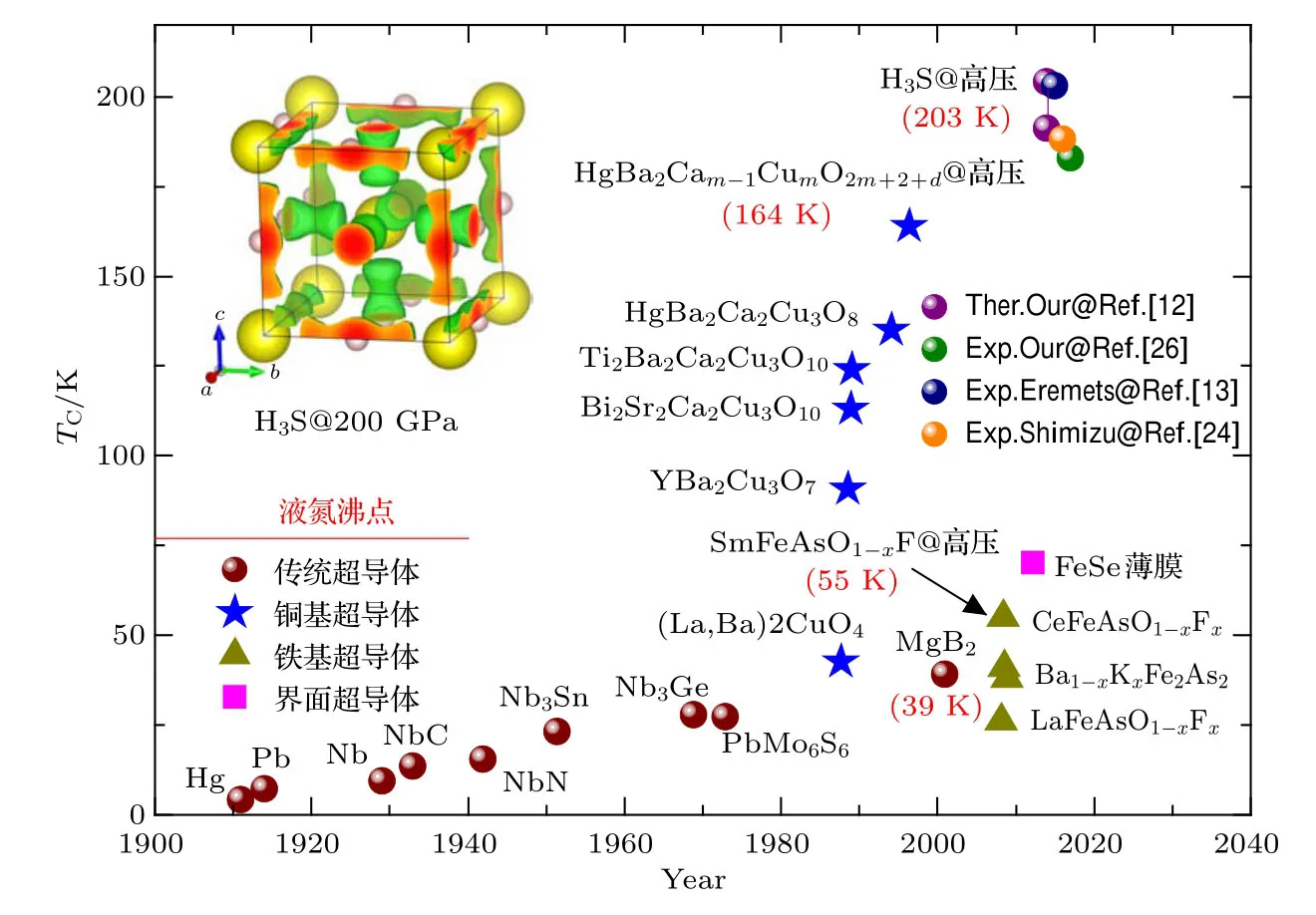
图1 超导材料的发现年代和临界温度 圆球代表传统超导体,五角星代表铜基超导体,三角形代表铁基超导体,方形代表界面超导体Fig.1.Discovery time of superconductors and critical temperature.Ball denotes conventional superconductor.Star denotes copper-based superconductor.Triangle denotes iron-based superconductor.Square denotes interface superconductor.
根据BCS理论,材料的超导转变温度与其德拜温度成正比,而德拜温度又与物质的质量成反比,因此可以预言自然界中最轻的元素氢可能具有很高的超导转变温度.但是固态氢在常压下为分子晶体,氢分子内存在很强的共价键,并且是绝缘体,不可能成为超导材料.为了实现超导,需借助压力等外界条件.高压能够非常有效地缩短原子间距离、增加相邻电子轨道重叠,进而改变原子(分子)间的相互作用和电子结构,形成常规条件下难以形成的具有新结构与新性质的高压新相.早在1935年,W igner和Huntington[15]就提出氢分子晶体在高压下转变成原子晶体,呈现金属状态,即金属氢.1986年,康奈尔大学的Ashcroft[16]认为金属氢极有可能是室温超导体.但是,目前实验压力已经达到388万大气压(GPa)左右,仍然没有获得氢金属化的直接证据[17].随着对金属氢这个重大物理问题研究的不断深入,人们开始寻找氢金属化的新途径.2004年,Ashcroft[18]又提出在富氢化合物中的非氢元素与氢元素之间存在相互作用,从而会对氢的子晶格产生化学预压作用,这类体系比纯氢更容易金属化,是潜在的高温超导体,有望成为超导家族的新成员—–氢基超导体.
不可否认,实验上确定高压下富氢化合物的结构、金属化及超导电性存在一定的困难,理论在这个领域的研究走在了前沿,并做出了很多重要的工作.理论上最常用的计算超导转变温度Tc的方法是Allen-Dynes修正后的McMillan方程
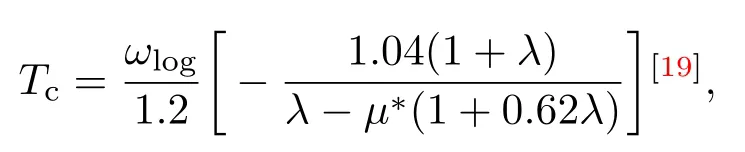
在这个方程中λ表示电声耦合常数;ωlog表示声子振动频率的对数平均值,其与超导转变温度Tc成正比;µ∗表示屏蔽库仑势,它是一个经验参数,对于富氢化合物通常选择0.10—0.15.从这个方程可知电声耦合常数λ、声子振动频率的对数平均值ωlog和屏蔽库仑势µ∗这三个参数共同控制超导转变温度Tc.
由于氢原子只有一个电子,兼具碱金属和卤素氧化性双重特性,高压下与其他元素形成富氢化合物时,会形成复杂的多中心电子键,从而表现出各种不同的晶体结构及性质.根据氢原子成键特征,可以把高压下的富氢化合物大致分为三类:含有原子氢的富氢化合物、含有H2分子单元的富氢化合物和含有H3分子单元的富氢化合物.对于含有原子氢的富氢化合物,根据非氢元素与氢元素的相互作用,又分为共价型富氢化合物和离子型富氢化合物,在这类富氢化合物中,氢原子表现出金属氢的特征,而非氢元素使得这个具有金属氢特征的结构在较低的压力下稳定存在,并具有较高的超导转变温度.对于含有H2分子单元的富氢化合物中,非氢元素与氢元素之间的相互作用变得比较复杂,而且很多富氢化合物在高压下都会出现H2分子单元.对于含有H3分子单元的富氢化合物,根据非氢元素的电负性,表现出线性的H阴离子和三角形的H阳离子,如与碱金属Rb形成了[Rb]+[H3]−,与卤族元素Cl形成[H3]+[Cl]−[H2].本文介绍这几类新型富氢化合物在高压下的晶体结构、稳定性、原子之间的相互作用、金属化及超导电性.
2 含有原子氢的富氢化合物
2.1 共价型富氢化合物
典型的共价型富氢化物包括硫氢化合物、硒氢化合物以及硼氢化合物等[12,20−22].2014年,我们课题组首次理论预测了新型氢化物H3S在高压下形成具有金属特性的立方相,空间群为Im-3m,如图2(a)所示.首次预言其在200 GPa的超导转变温度为191—204 K,突破200 K,并且Tc随压力的增大而单调降低[12],如图2所示.进一步明确了产生H3S晶体的两个主要途径:3H2S→2H3S+S,2H2S+H2→ 2H3S,即,可以对硫化氢直接加压到43万大气压以上就能获得[20],也可以通过硫化氢加氢在更低的压力(3.5 GPa)下获得[20,23].并且H3S晶体能稳定存在到300 GPa,其他氢含量更高的H4S,H5S和H6S化合物都不能稳定存在.2015年,德国马克斯-普朗克研究所的Eremets课题组[13]通过电学测量、迈斯纳效应测量和氢同位素效应测量,将H2S样品加压到155 GPa以上,发现在温度达到203 K(−70◦C左右)时变成超导体(图2),并且Tc也是随着压力的增大而减小,与我们前期理论预测值符合[12],认为高的超导温度来源于H3S,证实了我们的理论预言.最近,由日本大阪大学Shimizu课题组[24]通过高压同步辐射X射线衍射(XRD)和电阻测量证实了高温超导相来源于H3S,其结构就是我们理论预言的立方相[12],并且确认了H2S分解为H3S+S的机理[20],如图2(c)所示.Troyan等[25]利用同步辐射脉冲产生的核共振散射检测超导迈斯纳效应,发现153 GPa硫化氢向超导态转变且Tc为140 K.最近,我们课题组进行了高压硫化氢原位迈斯纳效应测量[26],在多个压力点得到了清晰的抗磁性信号,发现随着压力的增大Tc先增大后降低,在149 GPa下最高Tc为183 K,为H3S的高温超导电性提供了极其重要的实验证据,如图3(d)所示.
目前,实验和理论研究一致认为硫氢化合物在高压下200 K的超导转变温度来源于H3S的立方相Im-3m,并且确定高压下H2S分解为H3S+S的机理.但是目前还有两个问题不是很清楚:第一个问题,对于之前德国Eremets研究小组观测到另一个低温的超导相(30—70 K)结构还没有一致结论,可能是未分解完的P 1-H2S[27],也可能是介于H2S和H3S之前的P 1-H5S2[28],理论预测这两个结构的Tc与实验测量值符合得都比较好;第二个问题,高压下H2S的分解产物不是很清楚,110 GPa以下除了H3S,还存在H4S3,H5S8,H3S5和HS2等其他化学计量比的硫氢化合物[29,30].对于这两个问题,还需要进行更多的实验和理论研究.
高压下H3S具有200 K左右的Tc值是目前报道的最高的超导转变温度.从电子结构上看,H—S之间杂化作用强,电子布居高,可认为H—S形成了强的极性共价键[12,31].类似于MgB2,体系存在强共价键的同时具有金属性质,H3S被认为是又一个具有共价金属性的传统超导体[31].H—S的杂化作用不仅使得H以独立原子形式分布在S框架中,还使得H3S体系费米面处电子态密度发生重构,费米能级附近形成几乎不受压力影响的van-Hove峰,保证了体系优异的金属性[32,33].从电声耦合相互作用角度分析,H3S中H原子对电声耦合常数的贡献为82%,S原子贡献为18%[12],可以看出H原子对超导电性起着关键作用.综上所述,我们可以把H3S看作氢的原子相,而S能够使得这个原子相在较低的压力下稳定存在.
之后,人们希望在与硫同族的H-Se和H-Te体系中发现一些较好的超导材料,对于H-Te体系我们将在后面介绍.类似H3S体系,H3Se在166 GPa以上也出现Im-3m结构[21],但Se较大的原子半径使得H3Se的中频段声子谱和高频段声子谱分离,H3Se的电声耦合常数较H3S有所下降,但是在200 GPa时仍可以有110 K的超导转变温度.除了H3Se和常压下就存在的H2Se,H-Se体系中可能还存在HSe(Tc~40 K)以及富Se的HSe2(Tc~5 K)[21],HSe中H—Se之间共价键强度减弱,声子谱较H3Se出现明显软化,电声耦合常数减小,降低了体系的超导转变温度[21].由于共价金属性的存在是上述体系出现高Tc值的主要因素,理论计算认为掺杂微量与S(Se)电负性相近且质量更轻的元素例如P(As),原本母体体系的Tc值会进一步提高[34].

图2 (a)H3S立方相的晶体结构;(b)硫化氢在150 GPa的磁化率随温度的变化(黑色点)[13];(c)硫化氢在150 GPa下的XRD衍射图谱[24];(d)理论预测和实验测量的硫氢化物的超导转变温度随压力的变化[12,13,24,26,27]Fig.2.(a)The crystal structure of cubic phase of H3S;(b)temperature dependence of the magnetization of su lfu r hyd ride at a pressure of 155 GPa in zero-field cooled(ZFC)and 20 Oe field cooled(FC)modes(b lack circles)[13];(c)XRD patterns of su lfur hyd ride at 150 GPa[24];(d)pressu re dependence of superconducting transtion temperature Tcof su lfu r hyd ride by theoretical pred icted and experimental measured[12,13,24,26,27].
对于B-H体系,除了常规的B2H6化合物外,在高压下也可能出现新型硼氢化合物[22].理论预测BH在50 GPa到153 GPa之间可以与B2H6共存,在153 GPa后B2H6(P 21/c)分解,生成BH(Ibam)和H2[22].Ibam-BH在168 GPa转变为P 6/mmm结构,具有很强的B—B和B—H共价键,其在175 GPa的Tc为14.1—21.4 K.当压力达到350 GPa后,原本不稳定的B2H6重新变得稳定,形成了正交结构Pbcn[35].在这个结构中H原子与B之间形成共价键,在费米面附近显示出非局域状态,使得体系成为良好的导体,为产生较大的电声耦合常数提供可能.理论预测在360 GPa时,Pbcn-B2H6超导转变温度可达到125 K[35].
在共价型氢化物HnXm中,H将不会以独立的方式存在,而是与X元素形成较强的H—X共价键,结构具有很明显的整体性.这种整体性使得体系的光学支振动模式都对应H—X键的振动而非H本身.由于成键,更多的电子分布在H的周围,使得在费米能级附近的电子能够更充分地感受到H的振动.从谱函数上看,这种整体性使得体系中对电声耦合有贡献的频率范围变大,电声耦合常数变大.加之体系同时具备金属性,在费米面附近有更多的电子,使得此类体系有着非常奇异的超导电性.
2.2 离子型富氢化合物
代表性的离子型富氢化合物有Si2H6,AlH3和GaH3.我们课题组首次提出Si2H6在高压下形成Pm-3m结构,Si原子占据简单立方格点位置,H原子位于面心位置,更加振奋人心的是理论上预测这个简单结构的乙硅烷(Si2H6)在275 GPa的超导转变温度Tc高达139 K[36].理论预测AlH3和GaH3在高压下形成了A15结构(空间群为Pm-3n),GaH3在压力达到120 GPa时Tc高达102 K,而AlH3理论预测的超导转变温度在110 GPa时只有24 K.造成如此大差异的原因主要有两方面:1)在对应的压力点,GaH3费米面处的电子态密度要比AlH3大,而较大的电子态密度通常被认为是产生较大超导转变温度的一个必要条件;2)AlH3的电声耦合主要是由布里渊区高对称点X附近的声子振动模主控的[37],而GaH3的电声耦合是由全空间振动模式引起的,这种差异可能导致GaH3的电声耦合常数要比AlH3大得多,从而导致GaH3的超导转变温度要比AlH3高.但是实验测量上直到164 GPa,都没有观测到AlH3的超导电性,而GaH3的超导电性还有待于实验的证实.
另外一类典型的离子型富氢化合物为碱土金属氢化物CaH6[38],MgH6[39]和过渡金属氢化物YH6[40],它们在高压下形成体心立方结构空间群为Im-3m.在这类富氢化合物MH6(M=Mg,Ca,Y)中有一个共同特点——M原子占据体心立方结构的格点位置,H原子位于立方结构的六个面,形成了一个空间笼状结构.对于MH6(M=Mg,Ca和Y),每个面存在4个H原子,氢原子之间通过弱共价键相连,它们的距离在300,150和120 GPa分别为1.1,1.24和1.31Å,类似于氢的原子相.理论上预测MH6(M=Mg,Ca和Y)电声耦合常数在300,150和120 GPa分别为3.29,2.69和2.93,超导临界温度分别达到263,235和264 K.遗憾的是尽管这类富氢化合物呈现出较高的超导转变温度,但是一直没有得到实验证明.
3 含有H2分子单元的富氢化合物
高压下很多富氢化合物中H—H的距离相比同等压力下的固态氢中氢分子内部的H—H距离要长,而H与H之间还存在很强的共价键相互作用,所以这种H对通常被定义为H2分子单元.H2分子单元在富氢化合物中比较常见的,据不完全统计在不同化学配比的氢化物中至少70多种高压结构包含H2分子单元(仅限于二元氢化物),这些氢化物主要包括LiHn[41],NaHn[42],KHn[43,44],RbHn[45],CsHn[46], MgH4[47], CaH4,12[38], SrH4[48,49],BaHn[50], InH3,5[51], GeH4[52], SnH4[53,54],PbH4[55],AsH8[56],SbH4[57],BiHn[58],TeHn[59],PoHn[60],H3,7Cl[61],HnI[62,63],SiH4(H2)2[64,65]和GeH4(H2)2[66]等.这里只介绍几种典型的含有H2单元的富氢化合物,如图3所示.
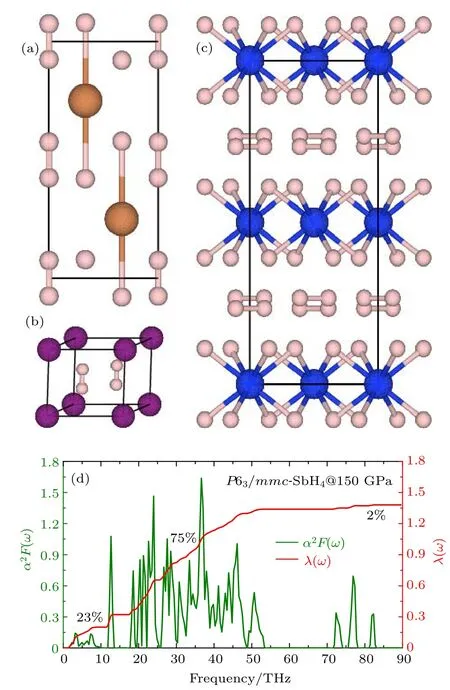
图3 (a)P 63/mmc-SbH4的晶体结构;(b)P 6/mmm-H4I的晶体结构;(c)C cca-SiH4(H2)2的晶体结构;(d)P 63/mmc-SbH4在150 GPa的Eliashberg光谱函数α2F(ω)和积分的电声耦合常数λ(ω)[58]Fig.3.The crystal structu re of(a)P 63/mmc-SbH4,(b)P 6/mmm-H4I,and(c)C cca-SiH4(H2)2;(d)the E liashberg phonon spectral function α2F(ω)and the electron-phonon integral λ(ω)of P 63/mmc-SbH4at 150 GPa[58].
除了H2分子单元,还存在多余的氢与非氢元素形成共价键或者离子键,如C2/c-GeH4[52],P 63/mmc-SnH4[54]和P 63/mmc-SbH4[57],它们在220,200和150 GPa的电声耦合常数分别为1.12,0.87和1.26,超导转变温度分别达到64,62和118 K.对于这三种氢化物,它们的声子谱主要被分为三部分:低频区主要是由非氢元素的振动引起的,中频区的振动主要与H的振动有关,而高频区域主要是由H2分子单元的振动导致的.H2分子单元的振动对电声耦合的贡献非常小,C 2/c-GeH4,P 63/mmc-SnH4和P 63/mmc-SbH4中H2分子单元对电声耦合的贡献仅仅达到了4%,3.7%和2%.P 63/mmc-SbH4的成键形式比较特殊,Sb原子与氢原子之间形成弱的共价键,与H2分子单元之间的作用主要是离子相互作用,如图3(a)所示.H2分子单元的存在有利于提高氢化物中声子振动频率的对数平均值(ωlog),在相应的压力点它们的数值分别达到了897,1135和1118.6 K.
高压下,SnH4,TeH4,H4I形成六角结构P 6/mmm[53,59,62],如图3(b)所示,在这类结构中所有的H全部形成H2分子单元,它们在120,170和120 GPa的电声耦合常数分别为1.20,1.46和0.50,超导转变温度分别为80,104和6 K.与其他含有H2分子单元的富氢化物类似,P 6/mmm-SnH4,P 6/mmm-TeH4和P 6/mmm-H4I中,H2分子单元的振动对电声耦合的贡献也非常小,分别为7%,5%和9.4%,但是它的出现被认为是有利于提高ωlog.P 6/mmm-SnH4是一个亚稳结构,出现了由费米面嵌套和科恩异常引起声子软化行为,声子软化有利于提高电声耦合强度,从而提高了其超导转变温度[53].Te-H体系没有形成类似于S-H[12,20]和Se-H[21]体系最稳定的MH3(M=S,Se)化学配比的氢化物,而是形成了P 6/mmm-TeH4这种离子型的氢化物[59],主要是因为Te元素的原子半径要比S与Se大得多,而且Te元素的电负性要比S和Se小.P 6/mmmH4I的超导转变温度很低,主要是由弱的电声子相互作用引起的.高压下H-I体系中还存在一个新的氢化物H2I,它存在两个高压相正交Pnma(100—246 GPa)和六角R-3m(246—300 GPa)[62].在Pnma相中,所有H形成H2分子单元,碘形成了单原子碘晶格.R-3m相中,H2分子单元消失,形成了原子相.进一步的电声相互作用计算表明Pnma和R-3m在240 GPa的Tc分别为3.8 K和33 K,发现原子相R-3m超导转变温度比它的前一个相Pnma(含有H2分子单元)提高了近8倍.
另外,实验上在6.8 GPa和7.5 GPa以上还分别合成了常压下难以形成的含H2分子更多的氢化物SiH4(H2)2和GeH4(H2)2[64,66],随后理论研究了它们在高压下的结构及超导电性.在这类氢化物中,MH4(M=Si,Ge)还保持其分子特征,MH4与H2分子单元之间主要是范德瓦耳斯相互作用.对于SiH4(H2)2的低压区(6.8 GPa),实验和理论提出多个可能结构:F-43m[64,67],I-4m2[68−70],Pmn21[69].另外理论还预测了高压金属结构P 1(125 GPa)和Ccca(248 GPa)[71],如图3(c)所示,Ccca在250 GPa的Tc为98—107 K,H2分子单元的振动对电声耦合的贡献仅有1%.对于GeH4(H2)2,理论也提出了很多高压结构,其中P 21/c结构稳定存在于220 GPa以上,理论预测它在250 GPa的Tc为76—90 K[72].
通过总结对比可以发现三个规律:1)含有H2分子单元氢化物的声子谱通常分为三个振动区域,低频区主要是由非氢元素的振动引起的,中频区的振动主要与H的振动有关,而高频区域主要是由H2分子单元的振动导致的;2)体系中的H2分子单元对体系的电声耦合强度λ的贡献非常小(小于10%),对电声耦合贡献最大的是中频区的振动;3)尽管H2分子单元对电声耦合的贡献很小,但是它的出现通常意味着体系具有较高的振动频率,从而导致一个较大的ωlog,能够提高超导转变温度Tc.
4 含有H3分子单元的富氢化合物
1911年,约瑟夫.汤姆逊[73]首次通过质谱仪发现了三氢阳离子H.1935年,库尔森[74]提出三氢阳离子具有平面正三角形空间构型的理论,其H—H键长为0.873Å.20世纪80年代,通过频率调制检测技术获得了H的光谱,随后的90年代早期则是在木星、土星和天王星的电离层中发现了H,2006年冈武史[75]宣布星际介质中普遍存在H.对于三氢H阴离子,理论提出直线型构型,并且认为H不稳定可能会分解成H2单元和H−,到20世纪90年代,第一性原理计算发现H在势能面上具有极小值[76],而一直到2003年才通过实验观测到它的存在[77,78].理论预测高压下在金属氢化物RbH3,5[45],CsH3[46],BaH6,8[50]和InH3[51],KH6[44]等出现了直线型的H分子单元,在H2F,H3F,H5F,H5Cl和H5Br中发现了三角形的H分子单元[61,79−81],但只有P 4/mmm-BaH6和R-3-InH3在高压下金属化,超导转变温度分别为38 K和40 K[50,51].
对于Rb-H体系[45],RbH5在15—220 GPa范围内最稳定,当压力大于220 GPa时,焓值最低的是RbH3,其稳定状态可以持续到250 GPa,且这两种配比的化合物都出现了直线型的H单元,如图4(a)所示.其中,RbH5-Cmcm结构在100 GPa下,H单元中H—H键的键长为0.91Å.RbH3存在两个高压相I41/amd和Cmmm,这两个相最大的不同之处在于:沿H分子轴线方向,前者的Rb+和H是交替出现的,而后者则是重复出现的.此外,在220 GPa压强以内,RbH5-Cmcm,RbH3-I41/amd和RbH3-Cmmm均未表现出金属性.与RbH3类似的是,CsH3以[H3]−[Cs]+的形式稳定存在于30—150 GPa压力范围内,低于40GPa时,焓值最低结构是Cmmm,40 GPa以上I41/amd相稳定,二者的焓值极为接近[46].CsH3-Cmmm结构中Cs+形成稍稍扭曲的三角形,并以AAA方式堆垛,H穿插在层间.对于H,层内与层间H—H键的键长分别为0.931和1.758Å.
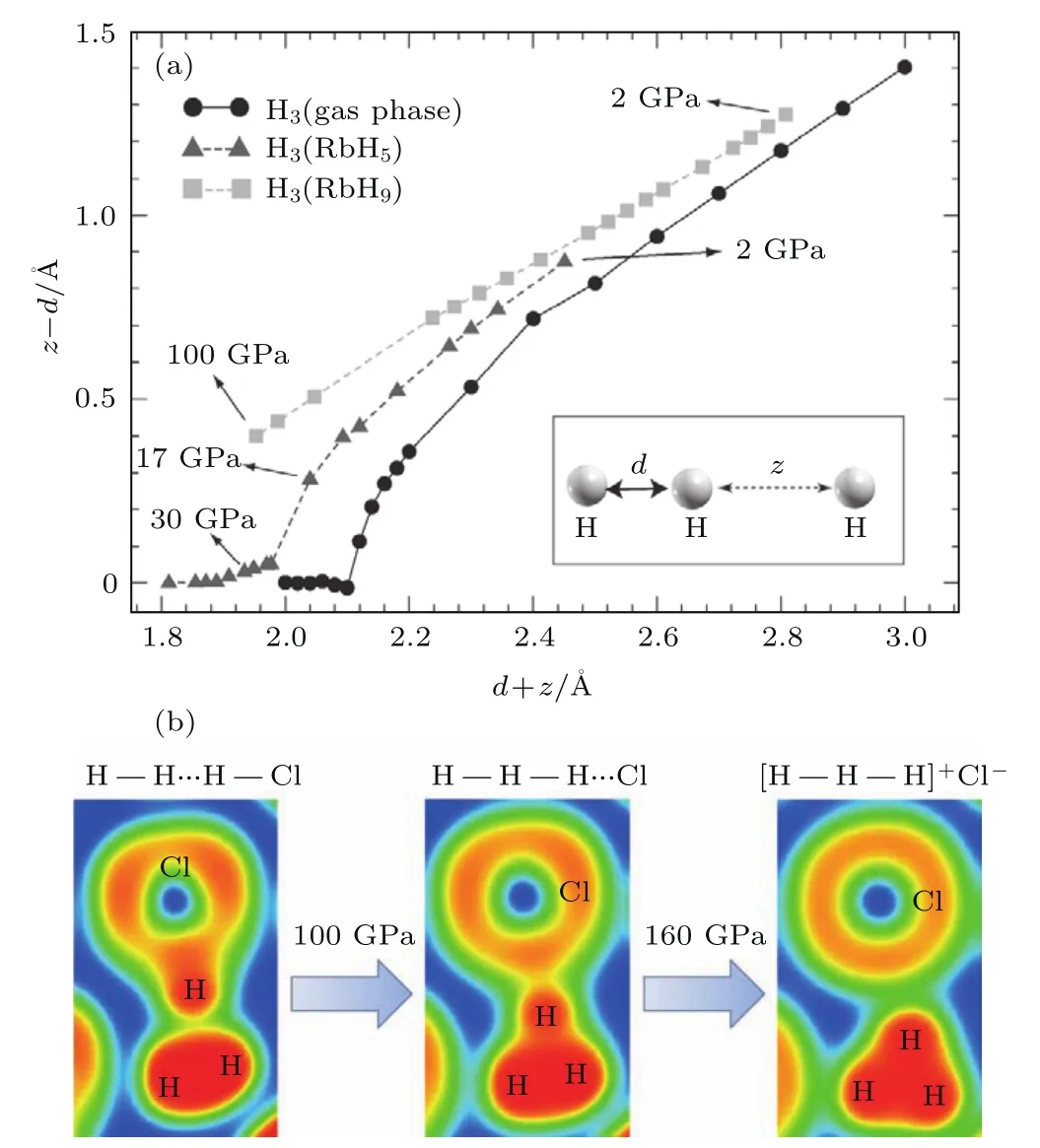
图4 (a)在气体相、lp-RbH9和lp-RbH5,H−...H2结构单元(插图)中z−d的值随z+d的值的变化[45];(b)Cc-H5C l结构的电子局域函数值随压力的变化[61]Fig.4.(a)D iff erence between the z and d distances in an H−...H2fragment(see inset)plotted versus their sumfor an isolated Hmolecu le in the gas phase,lp-RbH9and lp-RbH5[45];(b)electron localization function maps of Cc-H5C lwith increasing pressu re[61].
BaH6最稳定的结构是Fddd,以[H3]22−[Ba]2+的形式存在[50].其中,H3−在晶体中按人字形排列,70 GPa时,H分子内和分子间H—H键的距离分别为0.93和1.86Å.当压强增至80 GPa,BaH6开始变得不稳定,被BaH8所替代,继续加压至150 GPa,BaH6又重新落在凸包图上,此时出现了两个竞争相—–P 4/mmm和Imm2.与Fddd结构不同,Imm2结构中的H并不对称,其分子内H—H键的键长在150 GPa时分别为0.91和0.94Å,而P 4/mmm相中H的存在形式可以看成是H2分子和H−.在压力作用下,上述三种结构均可实现金属化,且BaH6-P 4/mmm在100 GPa时超导转变温度达到了30—38 K,电声耦合贡献主要来自H2单元和[H]−阴离子.
在In-H体系中[51],InH5-P 21/m结构在120 GPa时开始保持稳定直至233 GPa,进一步加压会导致InH5的分解,InH3-R-3结构将其取而代之,然后一直保持稳定到300 GPa.InH3在200 GPa时的R-3相中出现了直线型的H单元,In原子形成配位数为6的网状结构.其中,H3单元中最短的H—H键键长是0.872Å,In原子之间最短的距离为2.545Å.此外,通过电子局域函数和Bader分析得出In和H之间形成的是离子键,且电荷从In转移到H原子.通过计算其能带结构,发现InH3-R-3在200GPa下是金属化的,随后理论预测其超导转变温度为34.1—40.5 K,并且Tc随压力的增加而减小,主要原因是电声耦合常数λ和费米面处的电子态密度N(EF)随压力的增加而相对减小.
KH6存在两个稳定的高压相[44],70—166GPa范围内C 2/m相焓值最低,当压强大于166GPa时,C2/c结构开始变得稳定,一直持续到300 GPa.其中,C 2/m相中不仅有H2分子单元,还出现了直线型的H3分子单元,H2和H3单元共同组成一维网状结构.由于钾原子对氢原子的化学预压缩作用,使得电子从K转移到H,甚至部分H2单元解体,从而形成H3单元.100 GPa时,H3单元中H原子之间的距离分别为0.85和1.00Å.当压强增至166 GPa时,H3结构单元解离形成H2单元,架构成一维网状结构,并与K原子一起层层堆垛,这就是新的C2/c结构.通过电子结构计算,发现C2/m相为绝缘体,而C2/c相在其稳定的压强范围内是一种金属结构.对于C 2/c相,2个H原子成键,组成一维导电链结构,并且理论预测其在166 GPa的超导转变温度为58.66—69.84 K.
HBr与H2S类似,高压下变得不稳定,在64GPa会分解为新型溴氢化物H2Br和单质Br[79].与H5Cl相似,在H5Br中也出现了[H3]+[Cl]−[H2]形式,在103—140 GPa范围内以Cc结构存在(与H5Cl-Cc等结构),之后转变为能量更低的Pmn21结构.在140 GPa时,H5Br-Pmn21结构中的H3+分子键长分别为0.903,0.903和0.911Å.另外富氢化合物H2Br,H3Br,H4Br和H7Br在高压下也能稳定存在,都存在H2分子单元.H2Br以C 2/c结构稳定存在于30—180 GPa,在这个结构中包含两个HBr分子链和一个H2分子单元,它们之间靠范德瓦耳斯力结合形成了主客结构.之后变得不稳定,一直到240 GPa才以Cmcm结构稳定存在,理论预测其在240 GPa的Tc为12.1 K.与H—Cl化合物不同,在H-Br化合物中出现了H4Br,以P 63/mmc结构稳定存在于240—300 GPa,理论预测其在240 GPa的Tc为2.4 K.
与HnF和HnCl不同,高压下的H-I化合物中没有发现H3+分子单元.HI大约在6.7 GPa就会分解为元素单质H2和I2,在高压下又形成了含氢量较高的化合物H2I,H4I和H5I,而富碘的化合物HI2和HI3不能稳定存在[62],H2I和H4I的高压结构及超导电性已在前面介绍.
5 结论与展望
本文对高压下几种典型的含有原子氢、H2分子单元和H3分子单元的富氢化合物在晶体结构、稳定性、原子间相互作用、金属化、超导电性等方面进行了简单的介绍.通过对比可以发现,含有原子氢的富氢化合物在高压下普遍具有较高的超导转变温度,如实验和理论都证实Im-3m-H3S在高压下的Tc高达200 K,理论预测Im-3m-MH6(M=Mg,Ca,Y)在300,150和120 GPa的Tc分别达到263,235和264 K,Pm-3m-Si2H6在275 GPa高达139 K,Pm-3n-GaH3在120 GPa时Tc为102 K.对于含有H2分子单元的富氢化合物,有些超导转变温度较高,如P 63/mmc-SbH4,在150 GPa的Tc达到118 K,Ccca-SiH4(H2)2在250 GPa的Tc为107 K,与之形成对比的是,某些氢化物如P 63/mmc-H4I的Tc较低,在120 GPa只有6 K,H2分子单元在这类氢化物中扮演什么角色还需要继续深入研究.目前来看,含有H3分子单元的富氢化合物不具备较高的超导转变温度.另外,高压下富氢化合物的高对称性结构都呈现出非常高的超导转变温度,尤其是立方相结构,例如六角的P 63/mmc-SbH4和正交Ccca-SiH4(H2)2在高压下的Tc突破了100 K,而体心立方Im-3m-H3S和Im-3m-MH6(M=Mg,Ca,Y)在高压下的Tc都突破了200 K.早在20世纪70年代,著名的超导材料专家Matthias根据已知的金属及合金的超导转变温度的特点总结了6个规律,即著名的Matthias rules[82,83],尽管现在看来这几条规律都已经被打破,但是其中有一条“high symmetry isgood,cubic symmetry is best”对于富氢化合物来说还是适用的.总之,最近理论和实验发现高压下硫氢化物的超导转变温度为200 K,这给高压富氢化合物的研究注入了新的力量,期望以后总结经验规律,设计或者探索出可能具备更高超导转变温度的氢基超导体.
[1]Mazin II2015Nature525 40
[2]Bednorz J G,Müller KA1986Z.Physik.B64 189
[3]ZhaoZ X,Chen L Q,Cui C G,Huang Y Z,Liu J X,Chen G H,Li S L,GuoS Q,He Y Y 1987Chin.Sci.Bu ll.32 177(in Chinese)[赵忠贤,陈立泉,崔长庚,黄玉珍,刘锦湘,陈赓华,李山林,郭树权,何业冶 1987科学通报32 177]
[4]ZhaoZ X,Chen L Q,Yang Q S,Huang Y Z,Chen G H,Tang R M,Liu G R,Cui C G,Chen L,W ang L Z,GuoS Q,Li S L,Bi J Q 1987Chin.Sci.Bu ll.32 412(in Chinese)[赵忠贤,陈立泉,杨乾声,黄玉珍,陈赓华,唐汝明,刘贵荣,崔长庚,陈烈,王连忠,郭树权,李山林,毕建清1987科学通报32 412]
[5]Hor P H,Meng R L,W ang Y Q,GaoL,Huang Z J,Bechtold J,Forster K,Chu C W 1987Phys.Rev.Lett.58 1891
[6]GaoL,Xue Y Y,Chen F,X iong Q,Meng R L,Ramirez D,Chu C W,Eggert J H,MaoHK1994Phys.Rev.B50 4260
[7]Nagamatsu J,Nakagawa N,Muranaka T,Zenitani Y,Akimitsu J 2001Nature410 63
[8]Kamihara Y,W atanabe T,HiranoM,HosonoH2008J.Am.Chem.Soc.130 3296
[9]Chen X H,W u T,W u G,Liu R H,Chen H,Fang D F 2008Nature453 761
[10]Chen G F,Li Z,W u D,LiG,Hu W Z,Dong J,Zheng P,LuoJ L,Wang N L 2008Phys.Rev.Lett.100 247002
[11]Ren Z A,Lu W,Yang J,Y iW,Shen X L,Zheng C,Che G C,Dong X L,Sun L L,Zhou F,ZhaoZ X 2008Chin.Phys.Lett.25 2215
[12]Duan D,Liu Y,Tian F,Li D,Huang X,ZhaoZ,Yu H,Liu B,Tian W,Cui T2014Sci.Rep.4 6968
[13]D rozdov AP,Eremets MI,Troyan IA,Ksenofontov V,Shy lin S I2015Nature525 73
[14]W ang Q Y,Li Z,Zhang W H,Zhang Z C,Zhang J S,Li W,D ing H,Ou Y B,Deng P,Chang K,W en J,Song C L,He K,Jia J F,Ji S H,W ang Y Y,Wang L L,Chen X,Ma X C,Xue Q K2012Chin.Phys.Lett.29 037402
[15]W igner E,Huntington HB1935J.Chem.Phys.3 764
[16]Ashcroft N W 1968Phys.Rev.Lett.21 1748
[17]Dalladay-Simpson P,Howie R T,G regoryanz E 2016Nature529 63
[18]Ashcroft N W 2004Phys.Rev.Lett.92 187002
[19]Allen P B,Dynes R C 1975Phys.Rev.B12 905
[20]Duan D,Huang X,Tian F,Li D,Yu H,Liu Y,Ma Y,Liu B,Cui T2015Phys.Rev.B91 180502
[21]Zhang S,W ang Y,Zhang J,Liu H,Zhong X,Song HF,Yang G,Zhang L,Ma Y 2015Sci.Rep.5 15433
[22]Hu C H,Oganov AR,Zhu Q,Q ian G R,Frapper G,Lyakhov AO,Zhou HY 2013Phys.Rev.Lett.110 165504
[23]Strobel TA,Ganesh P,Somayazu lu M,Kent P R C,Hemley R J 2011Phys.Rev.Lett.107 255503
[24]Einaga M,Sakata M,Ishikawa T,Shimizu K,Eremets MI,D rozdov AP,Troyan IA,HiraoN,Ohishi Y 2016Nat.Phys.12 835
[25]Troyan I,Gavriliuk A,Rüff er R,Chumakov A,Mironovich A,Lyubu tin I,Perekalin D,D rozdov AP,Eremets MI2016Science351 1303
[26]Huang X,W ang X,Duan D,Bertil.S,X in L,Huang Y,Li F,Zhou Q,Liu B,Cui T2016 arX iv:1610.02630[cond-mat.supr-con]
[27]Li Y,HaoJ,Liu H,Li Y,Ma Y 2014J.Chem.Phys.140 174712
[28]Ishikawa T,NakanishiA,Shimizu K,Katayama-Yoshida H,Oda T,Suzuki N 2016Sci.Rep.6 23160
[29]Li Y,W ang L,Liu H,Zhang Y,HaoJ,Pickard C J,Nelson J R,Needs R J,LiW,Huang Y,Errea I,Caland ra M,Mau ri F,Ma Y 2016Phys.Rev.B93 020103
[30]Goncharov AF,Lobanov S S,Kruglov I,ZhaoX M,Chen X J,Oganov AR,Konôpková Z,Prakapenka V B2016Phys.Rev.B93 174105
[31]Bernstein N,Hellberg C S,Johannes MD,Mazin II,Meh l MJ 2015Phys.Rev.B91 060511
[32]Papaconstantopou los D A,Klein BM,Meh l MJ,Pickett W E 2015Phys.Rev.B91 184511
[33]Quan Y,Pickett W E 2016Phys.Rev.B93 104526
[34]Ge Y,Zhang F,YaoY 2016Phys.Rev.B93 224513
[35]Abe K,Ashcroft N W 2011Phys.Rev.B84 104118
[36]Jin X,Meng X,He Z,Ma Y,Liu B,Cui T,Zou G,MaoHK2010Proc.Natl.Acad.Sci.USA107 9969
[37]KimD Y,Scheicher R H,Ahu ja R 2008Phys.Rev.B78 100102
[38]W ang H,John S T,Tanaka K,Iitaka T,Ma Y 2012Proc.Natl.Acad.Sci.USA109 6463
[39]Feng X,Zhang J,GaoG,Liu H,W ang H2015RSC Adv.5 59292
[40]Li Y,HaoJ,Liu H,Tse J S,W ang Y,Ma Y 2015Sci.Rep.5 9948
[41]Zurek E,Hoffmann R,Ashcroft N W,Oganov AR,Lyakhov AO2009Proc.Natl.Acad.Sci.USA106 17640
[42]Baettig P,Zu rek E 2011Phys.Rev.Lett.106 237002
[43]Hooper J,Zu rek E 2012J.Phys.Chem.C116 13322
[44]Zhou D,Jin X,Meng X,BaoG,Ma Y,Liu B,Cui T2012Phys.Rev.B86 014118
[45]Hooper J,Zu rek E 2012Chem.A:Europ.J.18 5013
[46]ShampA,Hooper J,Zurek E 2012Inorg.Chem.51 9333
[47]Lonie D C,Hooper J,Altintas B,Zurek E 2013Phys.Rev.B87 054107
[48]Hooper J,Terpstra T,ShampA,Zu rek E 2014J.Phys.Chem.C118 6433
[49]W ang Y,W ang H,Tse J S,Iitaka T,Ma Y 2015Phys.Chem.Chem.Phys.17 19379
[50]Hooper J,Altintas B,ShampA,Zu rek E 2013J.Phys.Chem.C117 2982
[51]Liu Y,Duan D,Tian F,Liu H,W ang C,Huang X,Li D,Ma Y,Liu B,Cui T2015Inorg.Chem.54 9924
[52]GaoG,Oganov AR,Bergara A,Martinez-Canales M,Cui T,Iitaka T,Ma Y,Zou G 2008Phys.Rev.Lett.101 107002
[53]Tse J S,YaoY,Tanaka K2007Phys.Rev.Lett.98 117004
[54]GaoG,Oganov AR,Li P,Li Z,W ang H,Cui T,Ma Y,Bergara A,Lyakhov AO,Iitaka T,Zou G 2010Proc.Natl.Acad.Sci.USA107 1317
[55]Zaleski-E jgierd P,Hoffmann R,Ashcroft N W 2011Phys.Rev.Lett.107 037002
[56]Fu Y,Du X,Zhang L,Peng F,Zhang M,Pickard C J,Needs R J,Singh D J,Zheng W,Ma Y 2016Chem.Mater.28 1746
[57]Ma Y,Duan D,Li D,Liu Y,Tian F,Huang X,ZhaoZ,Yu H,Liu B,Cui T2015 arX iv:1506.03889[condmat.supr-con]
[58]Ma Y,Duan D,Li D,Liu Y,Tian F,Yu H,Xu C,ShaoZ,Liu B,Cui T2015 arX iv:1511.05291[cond-mat.suprcon]
[59]Zhong X,W ang H,Zhang J,Liu H,Zhang S,Song HF,Yang G,Zhang L,Ma Y 2016Phys.Rev.Lett.116 057002
[60]Liu Y,Duan D,Tian F,W ang C,W u G,Ma Y,Yu H,Li D,Liu B,Cui T2015RSC Adv.5 103445
[61]Duan D,Huang X,Tian F,Liu Y,Li D,Yu H,Liu B,Tian W,Cui T2015J.Phys.Chem.A119 11059
[62]Duan D,Tian F,Liu Y,Huang X,Li D,Yu H,Ma Y,Liu B,Cui T2015Phys.Chem.Chem.Phys.17 32335
[63]ShampA,Zu rek E 2015J.Phys.Chem.Lett.6 4067
[64]Strobel TA,Somayazu lu M,Hemley R J 2009Phys.Rev.Lett.103 065701
[65]W ang S,MaoHK,Chen X J,MaoW L 2009Proc.Natl.Acad.Sci.USA106 14763
[66]Strobel TA,Chen X J,Somayazu lu M,Hemley R J 2010J.Chem.Phys.133 164512
[67]Chen X Q,W ang S,MaoW L,Fu C L 2010Phys.Rev.B82 104115
[68]Michel K,Liu Y,Ozolins V 2010Phys.Rev.B82 174103
[69]Li Y,GaoG,Li Q,Ma Y,Zou G 2010Phys.Rev.B82 064104
[70]YaoY,Klug D D 2010Proc.Natl.Acad.Sci.USA107 20893
[71]Li Y,GaoG,X ie Y,Ma Y,Cui T,Zou G 2010Proc.Natl.Acad.Sci.USA107 15708
[72]Zhong G,Zhang C,Chen X,Li Y,Zhang R,Lin H2012J.Phys.Chem.C116 5225
[73]Thomson J J 1911Philos.Mag.21 225
[74]Cou lson C A1935Math.Proc.Cambridge Philos.Soc.31 244
[75]Oka T2013Chem.Rev.113 8738
[76]Stärck J,Meyer W 1993Chem.Phys.176 83
[77]W ang W,Belyaev AK,Xu Y,Zhu A,X iaoC,Yang X F 2003Chem.Phys.Lett.377 512
[78]Golser R,Gnaser H,Ku tschera W,Priller A,Steier P,W allner A,Čížek M,Horáček J,DomckeW 2005Phys.Rev.Lett.94 223003
[79]Duan D,Tian F,Huang X,LiD,Yu H,Liu Y,Ma Y,Liu B,Cui T2015 arX iv:1504.01196[cond-mat.supr-con]
[80]W ang Z,W ang H,Tse J S,Iitaka T,Ma Y 2015Chem.Sci.6 522
[81]Zeng Q,Yu S,Li D,Frapperb G,Oganov AR 2015 arX iv:1508.01395[cond-mat.mtrl-sci]
[82]Pickett W E 2001Physica B296 112
[83]Mazin II2010Nature464 183
PACS:61.50.Ks,74.70.Ad,74.62.FjDOI:10.7498/aps.66.036102
Structu res and novel superconductivity of hyd rogen-rich compounds under h igh pressu res∗
Duan De-Fang Ma Yan-Bin ShaoZi-Ji Xie Hui Huang Xiao-Li Liu Bing-Bing Cui Tian†
(State Key Laboratory of Superhard Materials,College of Physics,Jilin University,Changchun 130012,China)(Received 16 November 2016;revised manuscript received 3 December 2016)
Metallic hyd rogen can be realized theoretically at high pressure,which suggests that it will be a room-temperature superconductor due tothe high vibrational frequencies of hyd rogen atoms.However,themetallic state of hydrogen is not observed in experiment at upto388GPa.Scientistshave been exploring variousnewways toachieve hyd rogenmetallization.Hydrogen-rich compounds can bemetallized atmuch lower pressures because of chemical pre-compression.Moreover,because such materials are dominated by hyd rogen atoms,some novel properties can be found aftermetallization,such ashighTcsuperconductivity.Therefore,hyd rogen-rich compoundsare potentialhigh-temperature superconductors,and thismethod is alsobelieved tobe an eff ective way tometalize hydrogen,which has aroused signifi cant interest in lots of fields,such as physics,material science,etc.In a word,hydrogen-rich compounds are expected tobecome a newmember of superconductor family:hyd rogen-based superconductor.Very recently,the theoretical prediction and the successful experimental discovery of high-temperature superconductivity at 200 Kin a su lfur hydride compound at high pressure have set a record,which inspired further eff orts tostudy the superconductivity of hyd rogen-rich compounds.The present reviewfocuses on crystal structures,stabilities,interaction between atoms,metallization,and superconductivity of several typical hyd rogen-rich compounds at high pressures.Furthermore,higherTcsuperconductors can be expected tobe found in hyd rogen-rich compounds in the future.
high pressure,hydrogen-rich compounds,crystal structure,superconductivity
10.7498/aps.66.036102
∗国家自然科学基金(批准号:51632002,11674122,51572108,11204100,11504127,11634004)和教育部长江学者和创新团队发展计划(批准号:IRT_15R 23)资助的课题.
†通信作者.E-mail:cuitian@jlu.edu.cn
*Project supported by the National Natu ral Science Foundation of China(G rant Nos.51632002,11674122,51572108,11204100,11504127,11634004)and the Programfor Changjiang Scholars and Innovative Research Teamin University of Ministry of Education of China(G rant No.IRT_15R 23).
†Corresponding author.E-mail:cuitian@jlu.edu.cn
猜你喜欢
山东冶金(2022年4期)2022-09-14
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
科学与财富(2021年33期)2021-05-10
科学Fans(2019年2期)2019-04-11
金属世界(2018年4期)2018-08-11
环境保护与循环经济(2017年12期)2017-07-11
原子与分子物理学报(2014年4期)2014-02-28
故事作文·低年级(2009年11期)2009-12-10
