
利用二代测序数据判定SNP基因型
2017-08-01 11:00周宇荀肖君华
东华大学学报(自然科学版) 2017年3期
陈 科, 李 凯, 周宇荀, 肖君华
(东华大学 a.环境科学与工程学院; b.生物科学与技术研究所, 上海 201620)
利用二代测序数据判定SNP基因型
陈 科a, 李 凯b, 周宇荀b, 肖君华b
(东华大学 a.环境科学与工程学院; b.生物科学与技术研究所, 上海 201620)
通过将二代测序数据与连接酶检测反应(ligase detection reaction, LDR)对单核苷酸多态性 (single nucleotide polymorphism, SNP)基因分型的结果进行比对, 确定二代测序数据判定SNP基因型的经验性阈值.利用多重聚合酶链式反应(multiplex polymerase chain reaction, multiplex PCR)对91个样本进行19个SNP位点的扩增, 扩增的产物混匀纯化后在Ion torrent PGM仪器上进行二代测序.利用LDR技术对相应的SNP位点进行检测, 将其分型结果作为二代测序数据判定SNP基因型的标准, 确定了二代测序数据判定SNP基因型的阈值: 测序深度≥6X, 等位基因比率在15%~85%的位点为杂合子, 在范围之外的为纯合子, 该阈值准确度达到99.6%; 针对等位基因频率分布在阈值边缘的数据, 结合聚类分析可将正确率提升至100%.研究结果为利用二代测序数据判定SNP基因型提供了一个准确、快捷和经验性的阈值与方案.
单核苷酸多态性(SNP);二代测序;多重聚合酶链式反应(multiplex PCR)
单核苷酸多态性(SNP)是一种与复杂疾病相关的重要遗传位标[1].通常利用SNP进行生物学分析, 如遗传结构分析、个体化用药和复杂疾病定位.在过去的30年, 新SNP的发现依赖于以聚合酶链式反应(PCR)和毛细管电泳(capillary electrophoresis, CE)为基础的一代测序技术, 而SNP分型方案则依赖于Taqman、 Sequenom、SnaPshot、连接酶检测反应(LDR)和基因芯片等技术[2].近10年来, 二代测序技术高速发展, 其通量的不断提高以及成本的急剧下降, 使得二代测序技术开始成为SNP分型不可或缺的武器[3-4].
伴随着重测序技术, 如杂交后测序[5]、微液滴PCR[6]、分子内探针[7]等技术的开发, 将二代测序平台结合重测序技术应用于SNP分型已成为二代测序应用的重要部分[8], 并逐渐取代传统方案在SNP发现和分型上的作用[9-10].与此同时, Thermo Fisher Scientific和Illumina等供应商也开始提供商业化的试剂盒,以满足研究人员同时针对上百个样本和上百个目标区段内的SNP位点进行分型, 为二代测序技术进行大规模样本SNP分型提供了可行性[11-12].
二代测序低廉的测序价格和超高的测序通量是进行SNP分型的巨大优势, 但是在海量数据面前, 如何准确地进行SNP分型, 将是一个巨大的挑战.利用二代测序数据进行SNP分型, 困难主要集中于两个方面.一方面是等位基因偏差导致的困难.在二倍体个体中, 杂合子SNP位点的每条等位基因比率应该是50%, 但是在目标区段捕获和文库制备过程中, 因为捕获效率和扩增效率差异, 导致每条等位基因比率偏离50%.因此, 对于低覆盖度的测序(平均每个位点测序深度<5X), 会导致杂合子中只有其中一条等位基因被测序, 出现假阴性的情况.在这种情况下, 进行准确的SNP分型是非常困难的[13].虽然提高测序深度可以有效解决杂合子两条等位基因测序丢失的问题, 但是过度提高测序深度仍然无法彻底解决SNP分型错误的问题[14].另一方面是测序错误导致的困难.在二倍体个体中, 纯合子SNP位点的两条等位基因的基因型是相同的, 但是由于测序错误会导致纯合子的测序结果呈现出杂合子的可能性.随着测序深度的提高, 假阴性的可能性急剧下降, 但是测序错误经过累加, 使得纯合子判读越来越困难, 假阳性的比例升高.同时, 针对大规模平行测序时, 增加测序深度会导致成本急剧增加.因此, 提出一个准确、适当的SNP分型阈值, 是利用二代测序对大规模样本进行SNP分型时的重要补充.
本文利用多重PCR对91个样本进行19个SNP位点的扩增, 将PCR产物混合纯化后在Ion torrent PGM平台上进行二代测序.同时利用PCR-LDR技术对上述所有对应位点进行SNP分型, 结合二代测序数据进行验证, 以确定二代测序平台针对SNP分型的经验性阈值.以LDR分型结果为标准, 与二代测序数据进行对比, 提出一个准确、经验、有效的二代测序数据进行SNP分型的阈值和方案.
1 材料与方法
1.1 DNA提取
91个人血样,由无锡市精神卫生中心提供.DNA提取采用AXYGEN(杭州爱思进生物技术有限公司, 中国)血基因组小量制备试剂盒, 操作步骤依说明书进行.以0.8%琼脂糖凝胶电泳和NanoDrop2000c型超微量分光光度计 (Thermo Fisher Scientific, 美国)确定DNA质量和浓度.
1.2 引物的设计
19个SNP位点的rs号和基因序列信息来源于美国国立生物技术信息中心(National Center for Biotechnology Information, NCBI).所有特异引物用Primer3在线软件(http: //frodo.wi.mit.edu/primer3/)设计, 特异引物的解链温度(tm)在55 ~65 ℃, GC含量在20%~80%.特异引物上游和下游分别添加通用接头序列: 上游通用序列5’-tgtaaaacgacggccagt-3’;下游通用序列5’-caggaaacagctatgacc-3’(见表1).为标记不同样本, 设计携带不同index序列的接头引物以标记不同样本, 91个样本分别由不同的接头引物标记.所有引物皆由上海翰宇生物科技有限公司合成.三轮PCR原理示意图如图1所示,特异引物包括两个部分: 特异序列(白色)和通用序列(黑色);接头引物由3部分组成: Ion torrent接头序列(灰色)、特异index序列(斜线)和通用序列(黑色).
1.3 三轮PCR
PCR产物均一性是评价多重PCR的重要指标, 利用三轮PCR技术保证每对引物均匀扩增.三轮PCR使用的特异引物浓度极低, 在反应中又进一步地稀释特异引物浓度, 因此可以将低浓度特异引物尽可能地耗尽, 减少不同位点间的竞争, 以增加不同位点间均一性.与此同时, 用于文库构建的每对接头引物具有相同的扩增通用序列, 可以保证不同样本扩增时PCR效率的一致性, 以增加不同样本产物间的均一性.

表1 19个SNP位点引物
如图1所示: 第一轮PCR, 添加低浓度特异引物扩增;第二轮PCR, 取部分第一轮PCR产物作为模板添加到第二轮PCR体系中, 不额外添加引物扩增, 尽可能将第一轮残余引物消耗完;第三轮PCR, 直接将第三轮PCR体系添加到第二轮体系中, 利用接头序列将所有PCR产物添加上测序接头.经过三轮PCR后, PCR产物可以直接作为二代测序文库.
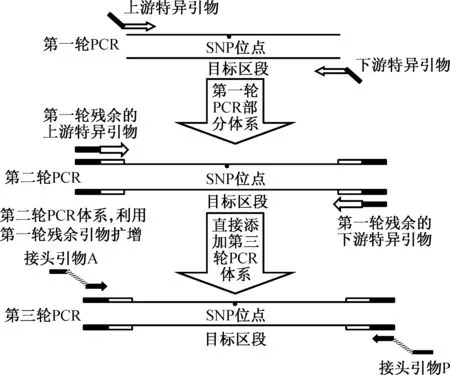
图1 三轮PCR原理示意图Fig.1 The schematic diagram of three-round PCR
1.3.1 第一轮PCR
PCR反应体系(10 μL)包括4.0 μL双蒸水, 1 μL 10×PCR Buffer(含100 mmol/L Mg2+), 0.8 μL dNTPs(2.5 mmol/L), 2 μL混合特异引物 (每条特异引物0.25 μmol/L), 1 U Hot Start DNA 聚合酶(5 U/μL) 和 2 μL DNA 模板(25 ng/μL).PCR反应程序: 94 ℃变性15 min, 94 ℃变性30 s, 60 ℃退火1 min, 72 ℃延伸30 s, 20个循环.
1.3.2 第二轮PCR
PCR反应终体系(10 μL)包括5 μL 双蒸水, 3 μL 第一轮PCR产物作为模板, 1.0 μL 10×PCR Buffer(含100 mmol/L Mg2+), 0.8 μL dNTPs(2.5 mM), 1 U Hot Start DNA 聚合酶(5 U/μL).PCR反应程序: 94 ℃变性15 min, 94 ℃变性30 s, 60 ℃退火1 min, 72 ℃延伸30 s, 40个循环.
1.3.3 第三轮PCR
第三轮PCR体系直接添加到第二轮反应体系中, 第三轮PCR反应体系(10 μL)包括7 μL 双蒸水, 1.0 μL 10×PCR Buffer(含100 mmol/L Mg2+), 0.8 μL dNTPs(2.5 mmol/L), 1 μL接头引物上下游引物(5 μmol/L), 1 U Taq DNA 聚合酶(1 μL) .PCR反应程序: 94 ℃变性15 min, 94 ℃ 变性30 s, 60 ℃退火1 min, 72 ℃延伸30 s, 15个循环;72 ℃补齐 10 min .
91个样本的PCR产物等量混合在一起, 振荡混匀.混匀的混合产物利用琼脂糖凝胶回收试剂盒(北京天根生化科技有限公司, 中国)纯化, 纯化后的PCR产物可直接用于后续测序反应.
1.4 Ion torrent PGM测序
纯化后的PCR产物于Ion torrent PGM测序仪(Thermo Fisher Scientific, 美国)上测序, 操作流程按照标准说明书进行.首先, 纯化后的PCR产物在OneTouch 2仪器上进行乳液PCR扩增, 通过乳液PCR扩增将测序片段富集在微珠上;乳液PCR扩增后的微珠由OneTouch 2 ES富集.富集的微珠结合318芯片(Thermo Fisher Scientific, 美国)和Ion PGMTMSequencing 200 Kit v2测序试剂盒在Ion torrent PGM仪器上测序, 测序流程严格按照说明书进行.
1.5 LDR
针对91个样本中19位点的LDR检测由上海翼和应用生物技术有限公司完成.
1.6 数据分析
二代测序数据经过base calling的原始数据通常包含3个部分: index序列、接头序列和特异序列.基于样本index序列的差异, 利用FASTX-Toolkit软件比对index序列, 根据index序列差异将原始数据中每个样本的数据区分, 在比对index序列时最多允许一个碱基的错配;匹配到每个样本的数据利用Cutadapt软件将接头序列去掉.运用序列比对软件BWA (v0.7.12)将去接头后的序列与SNP位点参考序列(NCBI)比对, 统计每个位点等位基因的reads数目, 计算其等位基因比率.
针对二代测序数据分型, 以LDR分型结果为标准, 即将LDR基因分型结果直接作为二代测序数据分型的结果.在此基础上, 将二代测序数据中位点的等位基因比率和LDR基因分型结果一一对应, 以确定二代测序数据中杂合子和纯合子等位基因比率阈值.
2 结果与分析
2.1 二代测序结果
针对91个样本, 对19个SNP位点进行SNP分型.以X代表扩增子的测序深度, 二代测序结果显示, 1 721个扩增子被成功捕获, 而且至少被测序一次(测序深度≥1X);SNP位点的捕获成功率为99.5%.扩增于测序深度分布图如图2所示.

图2 扩增子测序深度分布图Fig.2 Distribution of amplicons
由图2可知,1 721个扩增子的平均测序深度为136X, 96.2%扩增子的测序深度>平均测序深度的0.2倍;同时扩增子的测序深度呈现集中分布的趋势, 94.5%扩增子的测序深度集中在6X~300X (50倍差异).这种分布是利用二代测序技术对大规模样本进行SNP分型时测序深度分布的理想模型.该分布模型不仅保证了大多数扩增子有足够的测序深度, 同时避免了过多的高测序深度扩增子占用测序通量, 有效地节省了测序成本.在此测序深度分布模型的基础上, 结合LDR分型结果, 为二代测序数据进行大规模样本SNP分型提供一个准确的阈值和方案.
2.2 扩增子等位基因比率
二代测序数据显示有1 721个扩增子被成功测序, 将成功测序的扩增子等位基因比率进行统计.LDR分型结果显示,上述1 721个扩增子中杂合子为612个(35.6%)和纯合子为1 109个(64.4%).基于LDR技术的准确性[2], 将LDR的基因分型结果作为标准结果赋予二代测序数据: 利用二代测序数据中1 721个扩增子计算等位基因比率, 并直接选取LDR的分型结果, 为二代测序判定SNP基因型提供一个准确的阈值.扩增子等位基因比率如图3所示,以LDR技术对1 721个扩增子的分型结果为基础, 结合二代测序数据中对应扩增子等位基因的测序深度比值作图, 612个杂合子中, 610个(99.6%)杂合子等位基因比率在15%~85%.1 109个纯合子中, 1 105个(99.6%)纯合子等位基因比例在15%~85%之外.因此, 可以选取15%~85%作为判断杂合子和纯合子的阈值.
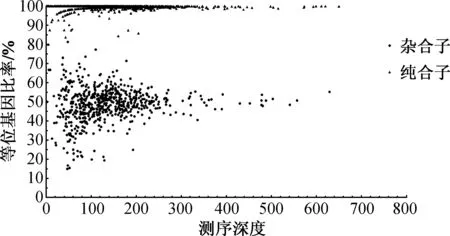
图3 扩增子等位基因比率Fig.3 Allelic ratio of amplicons
SNP位点等位基因比率分布如图4所示.由图4可知,94.5%的纯合子等位基因比率分布在98.0%~100%之外;89.0%的杂合子等位基因比率分布在40%~60%.杂合子等位基因比率随着测序深度的增加, 逐渐接近理想值50%, 说明随着测序深度的提升, 杂合子的分型越来越来越容易(见图3).与之相对的是纯合子分型, 纯合子的等位基因频率不随测序深度变化而转变, 说明对于低测序深度, 纯合子的判读更容易.
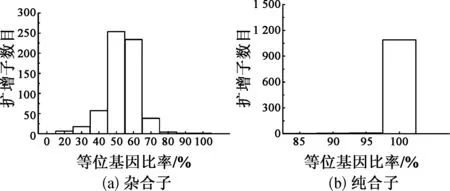
图4 SNP位点等位基因比率分布Fig.4 The allelic ratio distribution of amplicons
杂合子等位基因比率在15%~85%之外的2个数据中, 其中一个数据(rs140489)测序深度为2X, 偏差原因可能是测序深度过低, 导致等位基因只有一条被成功测序, 导致等位基因缺失[14];另外一个数据(rs3739470)测序深度为47X, 等位基因比率为14.9%, 该位点等位基因比率接近15%~85%阈值.类似的结果也发生在1 109个纯合子中, 纯合子等位基因偏差的4个数据中, 2个数据(rs140489和rs4842131)测序深度为5X, 偏差原因可能是测序深度过低, 随机测序错误引入导致等位基因比率变化过大;另外两个数据(rs5754217和rs3093024)的测序深度分别为46X和160X, 等位基因比率分别是83.7%和84.4%, 接近15%~85%阈值之外. 上述结果表明, 针对二代测序数据判定SNP基因型选取15%~85%作为阈值, 无论纯合子或者杂合子的准确性都达到了99.6%, 难以判断的数据只是过低的测序深度(<5X)和极度接近阈值边缘的数据, 显示了该阈值的准确性和适用性.
2.3 SNP位点等位基因比率
对19个SNP位点分型的二代测序数据进行统计分析,如2.2节描述, 610个(99.6%)杂合子的等位基因比率在15%~85%, 1 105个(99.6%)纯合子等位基因比率在15%~85%之外, 将上述纯合子和杂合子具体到每个SNP位点分析.SNP位点等位基因比率如图5所示.


图5 SNP位点等位基因比率Fig.5 The allelic ratio of SNP loci
由图5(a)可知,每个SNP位点的杂合子等位基因比率分布有偏差, 19个SNP位点中有16个(84%)SNP位点的杂合子偏差集中在40%~60%, 另外3个SNP位点在20%~80%.与之相对的, 19个SNP位点中纯合子无明显偏差(见图5(b)).不同SNP位点的杂合子等位基因比率有明显差异, 但是每一个SNP位点内等位基因偏差较小.其中最大偏差位点为rs140489, 从27.2%至100%, 相差72.8%;相对地, 最小偏差为位点rs3739470, 从19.3%至24.8%, 相差5.5%(见表2).

表2 SNP位点等位基因比率偏差
图5显示每个SNP位点内杂合子偏差和纯合子偏差有明显区别, 利用该差异并结合2.2节所描述的分型阈值,可以对极度接近阈值边缘的位点做进一步分析,以便提高SNP分型的准确性.如2.2节所述, 针对等位基因比率15%~85%的阈值, 612个杂合子有2个数据偏离. 其中一个数据为rs140489, 等位基因比率100%, 去除该数据后, 该SNP位点杂合子等位基因偏差范围从27.2%~100%降低到27.2%~66.7%, 偏差原因是位点测序深度过低(2X), 导致等位基因测序缺失;另外一个数据为rs3739470, 该数据测序深度为47X, 等位基因偏差为14.9%, 按照15%~85%的阈值, 该位点被错误地判断为纯合子.通过等位基因比率结合聚类分析可以纠正疑似位点的错误判读. SNP位点等位基因比率偏差如表2所示, 由表2可知, SNP位点rs3739470的纯合子偏差在98.1%~100%, 杂合子偏差在14.9%~24.9%, 可以判断14.9%的偏差仍为杂合子.同样的, 针对1 109个纯合子中偏离的4个数据, 其中两个数据测序深度为5X, 偏差原因是测序深度太低, 导致随机测序错误引入引起等位基因剧烈变化.另外两个(rs5754217和rs3093024)测序深度>40X,其杂合子偏差分别在39.5%~68.0%和33.3%~61.1%;而纯合子偏差分别是83.7%~100%和84.4%~100%. 按照2.2节所述阈值, 偏差83.7%和84.4%的位点为杂合子, 但结合表2中杂合子和纯合子的分布差异, 可以判断这两个点为纯合子.因此, 当测序深度≥6X, 利用15%~85%的阈值结合聚类分析, 对二代数据进行SNP分型的准确度可以达到100%.
采用二代测序技术对SNP进行分型时, 利用15%~85%的经验性阈值判断SNP是准确而便捷的, 但是杂合子位点等位基因比率有时会偏离但接近阈值, 原因是由于该SNP位点周围空间构象等问题导致引物结合偏好性不同, 或者SNP位点序列特异导致PCR扩增偏好, 从而引起等位基因偏好扩增[7, 15-16].这些SNP位点中杂合子等位基因比率会偏离阈值, 针对这些偏离但接近阈值的位点, 必须结合聚类分析才能进行准确的SNP分型.
3 结 语
本文利用三轮PCR针对91个样本进行19个SNP位点的扩增, 将扩增产物进行二代测序检测.测序结果显示1 721个扩增子被成功捕获.1 721个扩增子的基因型以LDR的基因分型结果作为标准结果, 同时利用二代测序数据计算1 721个扩增子的等位基因的比率, 两者数据结合分析确定了二代测序数据判定SNP基因型的阈值和方案.结果显示: 测序深度≥6X, 等位基因比率在15%~85%的为杂合子, 反之为纯合子, 其准确度为99.6%.但该阈值具有一定的局限性, 当杂合子或者纯合子的等位基因比率接近阈值边缘时难以准确判定, 解决方案是针对该位点进行聚类分析从而进行清晰的SNP分型.本文成功地提出了一个准确、经验性的针对二代测序进行SNP分型的阈值, 同时利用聚类分析方案作为SNP分型阈值的补充, 有效地提高了对易出错位点判读的准确率.本文研究结果是利用二代测序技术对大规模样本进行SNP分型应用的重要补充.
[1] SHAVRUKOV Y, SUCHECKI R, ELIBY S, et al. Application of next-generation sequencing technology to study genetic diversity and identify unique SNP markers in bread wheat from Kazakhstan[J]. BMC Plant Biology, 2014, 14: 258.
[2] SYVANEN A C. Accessing genetic variation: genotyping single nucleotide polymorphisms[J]. Nature Reviews Genetics, 2001, 2(12): 930-942.
[3] MARDIS E R. The impact of next-generation sequencing technology on genetics[J]. Trends in Genetics, 2008, 24(3): 133-141.
[4] SCHUSTER S C. Next-generation sequencing transforms today’s biology[J]. Nature Methods, 2008, 5(1): 16-18.
[5] GNIRKE A, MELNIKOV A, MAGUIRE J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing[J]. Nature Biotechnology, 2009, 27(2): 182-189.
[6] TEWHEY R, WARNER J B, NAKANO M, et al. Microdroplet-based PCR enrichment for large-scale targeted sequencing[J]. Nature Biotechnology, 2010, 28(2): 1025-1031.
[7] LEPROUST E. Target enrichment strategies for next generation sequencing[J]. Mlo Medical Laboratory Observer, 2012, 44(6): 111-118.
[8] MOKRY M, FEITSMA H, NIJMAN I J, et al. Accurate SNP and mutation detection by targeted custom microarray-based genomic enrichment of short-fragment sequencing libraries[J]. Nucleic Acids Research, 2010, 38(10): e116.
[9] LI R, LI Y, FANG X, et al. SNP detection for massively parallel whole-genome resequencing[J]. Genome Research, 2009, 19(6): 545-552.
[10] BOLAND J F, CHUNG C C, ROBERSON D, et al. The new sequencer on the block: Comparison of Life Technology’s Proton sequencer to an Illumina HiSeq for whole-exome sequencing[J]. Human Genetics, 2013, 132(10): 1153-1163.
[11] CONSORTIUM T 1 G P, ALTSHULER D L, DURBIN R M, et al. A map of human genome variation from population-scale sequencing[J]. Nature, 2010, 467(7319): 1061-1073.
[12] 王乐, 叶健, 白雪, 等. 二代测序技术及其在法医遗传学中的应用[J]. 刑事技术, 2015(5): 353-358.
[13] NIELSEN R, PAUL J S, ALBRECHTSEN A, et al. Genotype and SNP calling from next-generation sequencing data[J]. Nature Reviews Genetics, 2011, 12(6): 443-451.
[14] ROBASKY K, LEWIS N E, CHURCH G M. The role of replicates for error mitigation in next-generation sequencing[J]. Nature Reviews Genetics, 2014, 15(1): 56-62.
[15] QUINLAN A R, MARTH G T. Primer-site SNPs mask mutations.[J]. Nature Methods, 2007, 4(3): 192-192.
[16] MERTES F, ELSHARAWY A, SAUER S, et al. Targeted enrichment of genomic DNA regions for next-generation sequencing[J]. Briefings in Functional Genomics, 2011, 10(6): 374-386.
(责任编辑: 杨 静)
SNP Genotyping Based on Next Generation Sequencing Data
CHENKea,LIKaib,ZHOUYuxunb,XIAOJunhuab
(a. School of Environmental Science and Engineering; b. Institute of Biological Sciences and Biotechnology, Donghua University, Shanghai 201620, China)
The next generation sequencing data were compared with ligase detection reaction(LDR) data to determine the empirical cut-off thresholds of next generation sequencing data for single nucleotide polymorphism (SNP) calling. The 19 loci from 91 human genomic DNA were amplified with multiplex polymerase chain reaction(multiplex PCR). Then, all the amplicons were sequenced in a single run on Ion torrent PGM platform.With LDR genotyping data, the empirical cut-off thresholds of next generation sequencing data for SNP calling were that sequencing depth was ≥6X and heterozygote ratio was fell in 15%-85%. Application of this method was able to accurately determine 99.6% of SNPs, but was failed to judge the data closed edge of the cut-off thresholds. Combining with clustering analysis could solve this problem for increasing the accuracy to 100%. The results of research provided an accurate, fast and empirical threshold for the next generation sequencing for single nucleotide polymorphism calling.
single nucleotide polymorphism(SNP); next generation sequencing; multiplex polymerase chain reaction(multiplex PCR)
1671-0444 (2017)03-0370-07
2016-05-17
国家自然科学基金面上资助项目(31371257);上海市科委关键资助项目(14140900502)
陈 科(1988—),男,河南焦作人,博士研究生,研究方向为核酸检测新技术. E-mail: ck20dyj@163.com 肖君华(联系人),男,教授,E-mail:xiaojunhua@dhu.edu.cn
Q 446
A
猜你喜欢
数学物理学报(2022年1期)2022-03-16
南京农业大学学报(2020年4期)2020-07-28
莫愁(2019年34期)2020-01-01
伴侣(2019年7期)2019-07-25
莫愁·智慧女性(2019年12期)2019-06-01
消费导刊(2018年8期)2018-05-25
中国现代医学杂志(2016年19期)2016-11-09
中国惯性技术学报(2015年1期)2015-12-19
考试周刊(2015年53期)2015-09-10
