
水合单核铝到二聚铝形态的键合水分子活性变化研究
2018-01-04 22:57金晓艳王林玉钱佳佳蒋雪月
阜阳师范大学学报(自然科学版) 2017年4期
金晓艳,林 玲,王林玉,钱佳佳,蒋雪月
(阜阳师范学院 化学与材料工程学院,安徽 阜阳 236037)
水合单核铝到二聚铝形态的键合水分子活性变化研究
金晓艳,林 玲,王林玉,钱佳佳,蒋雪月
(阜阳师范学院 化学与材料工程学院,安徽 阜阳 236037)
采用密度泛函方法研究了水溶液中单核铝形态和二聚铝形态中键合水分子的活性。计算结果表明,单核铝发生聚合形成二聚铝形态导致键合水分子的活性明显增加。对单核铝而言,水解和氟离子取代均会增加水分子的活性,而对于二聚铝形态,由于羟桥和分子内氢键的出现,水解和氟离子取代对活性无明显影响。该结果为进一步揭示铝形态的水解聚合机制提供了重要的动力学参数。
密度泛函;单核铝;二聚铝;键合水分子活性
铝在地表中通常以各种铝氧矿物的形式存在,其在地壳中的含量仅次于氧元素和硅元素,是地壳中含量第一的金属元素。由于整个地球环境化学处于水溶液体系中,因此水合铝形态的化学性质就成为了地球化学和环境科学的重要研究领域[1]。此外,可溶性Al3+可以通过饮水、食物等方式进入生物体内,由于近年来酸雨的日益严重和工业废水的排放,地表水中过高浓度的可溶性铝已对动植物产生严重危害,因此,水合铝化学也同时成为了“铝毒”研究的重要基础[2]。在这种情况下,与元素周期表中相邻的其它元素硼、硅、镓、锗相比,铝化学受到的关注最多。
在水溶液体系中,具有高电荷和小体积的Al3+拥有较高的电荷密度,这一结构特点决定了Al3+与周围溶剂水分子间可产生强烈相互作用。大部分铝盐在pH<3的水溶液中以Al(H2O)63+的形态存在[3],随pH升高,这一形态会发生水解和聚合反应,除了产生一系列的水解单核铝形态之外,还会形成不同程度的聚合铝形态,如研究最为广泛的Keggin-Al13形态[4-7]。毫无疑问,这种水解聚合反应直接导致了水溶液中铝形态存在的复杂性。尽管到目前为止,水溶液中的各种铝形态仍未能完全弄清,但是对已知铝形态的键合水分子的活性研究却受到了学界很大的关注,因为这一性质决定了该铝形态参与其它各种化学反应的动力学行为。这种键合水分子的活性通常以17O NMR测定的该铝形态的水交换反应速率常数来表示。在早期的研究中,单核铝及其一级水解产物以及氟离子取代产物的键合水分子的活性均已通过这一方法测定,结果表明水解或氟离子取代均可明显增进其活性[8]。
二聚铝作为最简单的聚合铝形态,其水合形态近年已通过Al(OH)3合成[9],其键合水分子的活性直接决定了随后的聚合行为和其它聚合铝形态的形成。然而由于二聚铝形态在水溶液中的不稳定性,其水交换反应速率常数至今仍未能实验测定,在这种情况下,理论计算就成为目前解决这一难题的唯一方法。理论计算研究由于可以从分子水平对反应机理进行研究,经常作为实验技术的有力补充。目前对单核铝形态的理论计算已有较为充分的研究[10],由于有丰富的实验数据作为参照,合理的理论计算方法易于建立。若能在单核铝研究建立的理论基础上将理论计算应用于键合水分子活性完全未知的二聚铝形态,则既可以保证二聚铝形态研究结果的可靠性,又可了解从单核铝到二聚铝形态中的活性变化,从而为其它聚合铝形态键合水分子的活性研究提供理论指导。
在铝形态的实验研究中,OH-和F-是最常涉及的两种配体,OH-取代可直接代表pH对铝形态的影响,而pH的变化正是铝形态水解聚合的前提。此外,F-作为环境表界面中广为存在的一种形态,它与Al3+之间强烈的配位作用使得F-成为影响环境矿物表界面化学反应的一个重要因素。因此本文不仅采用密度泛函方法研究了单核铝和二聚铝形态中键合水分子的活性变化,而且在此基础上分别考察了OH-和F-取代对二者活性的影响。首先,通过模拟单核铝的水交换反应过程,获得决定其速率常数的活化能垒,随后,分别模拟OH-和F-取代产物的水交换反应过程,以探讨水解和氟化对键合水分子活性的影响,最后,在此基础上模拟二聚铝形态不同键合水分子位点的水交换反应过程,并分别探讨水解和氟离子取代对二聚铝形态键合水分子的活性影响,从而得出活性影响的对比结论。该结论为进一步揭示水合铝形态的水解聚合机制提供了动力学基础,也为实验探测铝氧矿物的表界面反应提供了可靠的理论依据。
1 计算方法
本文所有计算均采用Gaussian 09软件完成,所有团簇均在PBE0-D3[11]基础上进行气相优化,其中O原子和H原子采用了全电子基组6-311+G(d,p),Al原子采用LANL2DZ赝势,所有优化结构随后进行频率计算,以局域最小确定反应物和中间体,以唯一振动虚频确定过渡态。考虑溶剂效应对能量的影响,在所有气相优化构型的基础上进行SMD[12]计算,以获得反应过程中准确的吉布斯自由能变。
2 结果与讨论
2.1 单核铝键合水分子活性变化
图1给出了单核铝形态水交换反应过程,相应的结构参数的变化列于表1中。从图1可以看出,在反应物(R)中,键合水分子与铝离子形成了六配位的八面体结构,铝水键长约为1.949 Å,接近于实验值1.90 Å。接下来,其中一个键合水分子渐渐远离中心铝离子,这表示水交换反应开始发生,当这一铝水键长(Al-OH2(L))拉升至2.863 Å时,过渡态(TS)出现。随后,该水分子进一步远离中心铝离子,最终与其它两个键合水分子以氢键键合,形成了五配位的中间体(I),此时Al-OH2(L)键长达到了3.421 Å,表明这一水分子已完全进入第二水化层。整个过程中,离去水分子之外的其它水分子(即旁观水分子)的铝水键长(Al-OH2(S))明显减小,这是由于配位数的减少导致中心铝离子对周围配体的键合力作用增强而引起。从表2的能量数据可以得出,这一过程的活化吉布斯自由能垒约为84.6 kJ·mol-1,反应能量为79.5 kJ·mol-1,表明了五配位中间体(I)的不稳定性。
当Al(H2O)63+发生水解时,其中一个水分子配体脱去一个质子形成了羟基配体,此时Al(OH)

图1 Al(H2O)63+水交换反应过程 红色表示氧原子,白色表示氢原子,紫色表示铝离子,绿色表示离去水分子
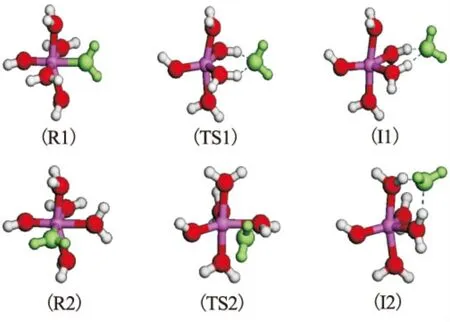
图2 Al(OH)(H2O)52+水交换反应过程 R1、TS1和I1分别对应于羟基对位水分子水交换反应的反应物、过渡态和中间体;R2、TS2和I2分别对应于羟基邻位水分子水交换反应的反应物、过渡态和中间体。
(H2O)52+团簇中出现了两种不同化学环境的水分子,分别是与羟基呈对位和邻位的水分子。从表1可以看出,对位水分子的铝水键长比邻位水分子略有增加。这两种水分子的水交换反应过程如图2所示,从中可以看出,这两个水分子在离去过程中,均通过与相邻两个键合水分子形成氢键而产生中间体。有趣的是,尽管二者铝水键长有~0.02 Å的差异,但是其活化能垒几乎完全相同,表明反应过程中水分子的离去速率并非完全由铝水键长控制。同时也可得出,单核铝形态一旦发生水解,其键合水分子的能垒几乎下降了一半,表明活性大大增强,这一点也与17O NMR实验结论一致[13],通常被认为由整个团簇表观电荷下降引起。
众所周知,对水合铝形态来说,F-很容易取代其中的键合水分子。其氟离子取代产物AlF(H2O)52+的水交换过程与Al(OH)(H2O)52+相似,区别仅在于其中的羟基被F-替代。同样,对位和邻位水分子的活性亦没有明显差异,均比Al(H2O)63+的能垒下降了~20 kJ·mol-1,表明了活性的增加。但值得注意的是,F-取代引起能垒下降幅度稍小于OH-,尽管 F-和 OH-的电荷均为-1,F 和 O 原子大小也较接近,因此正如文献所述[13],F和O之间电负性的差异应是这种活性变化的主要原因。
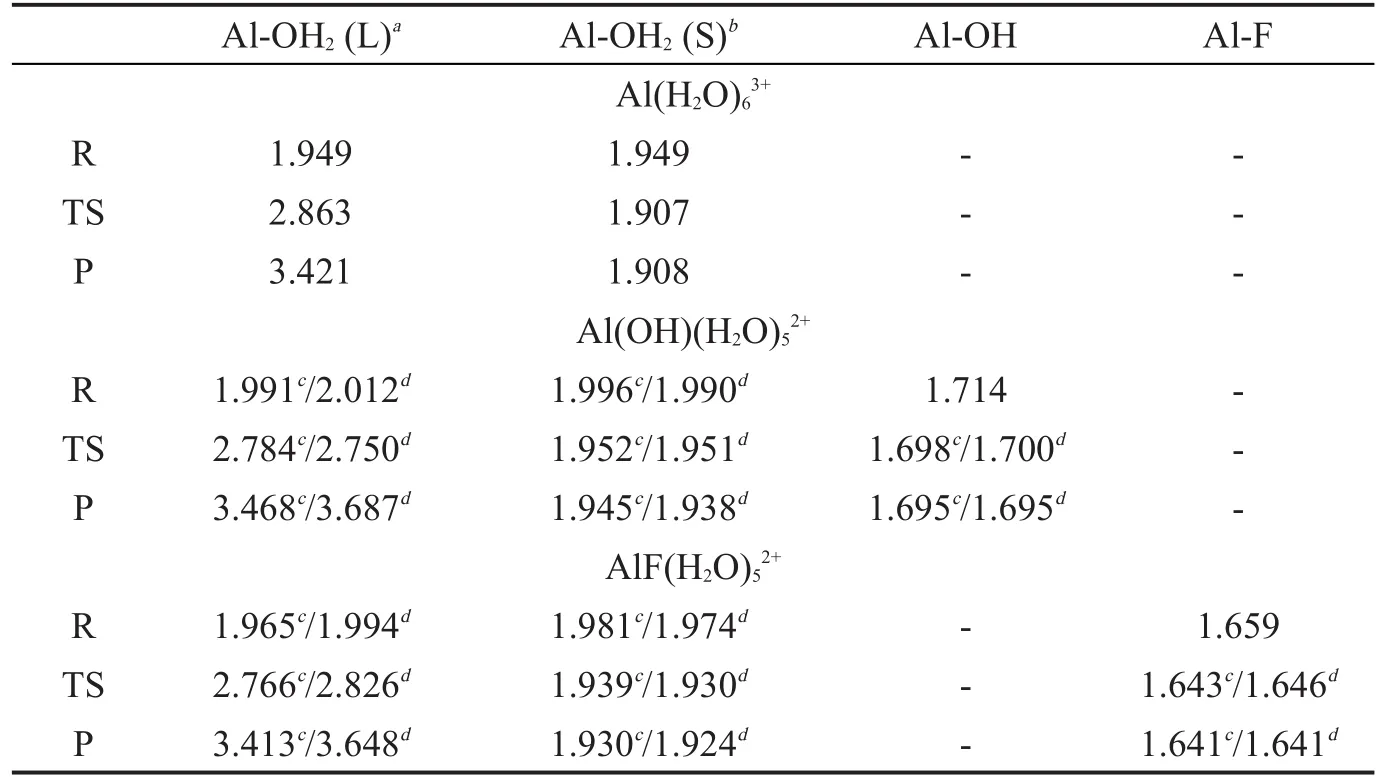
表1 单核铝形态水交换反应过程结构参数的变化/Å
与实验活化能垒相比较,除Al(H2O)63+高估了~12 kJ·mol-1外,Al(OH)(H2O)52+和 AlF(H2O)52+的计算能垒与实验值几乎一致。考虑到DFT计算的吉布斯自由能与实验值的差异通常在10~20 kJ·mol-1以内,说明本文所采用计算方法是合理的。
2.2 二聚铝形态键合水分子的活性变化
当单核铝发生聚合形成二聚铝形态之后,两个铝离子通过两个羟桥连接,该结构中含有两种不同化学环境的水分子,如图3所示,反应物R1中的赤道面水分子和R2中的轴向水分子。从表3列出的结构参数可知,轴向水分子的铝水键长大于赤道面水分子对应的铝水键长,但二者均高于单核铝形态的铝水键长,表明聚合引起整个结构向外伸展。就轴向水分子的水交换反应而言,该水分子配体通过与另一铝离子上的轴向水分子作用而渐渐远离中心铝离子,经历过渡态之后形成了一个五配体的中间体。而赤道面水分子通过与相邻的两个水分子配体作用渐渐离开中心铝离子,并最终形成中间体。从表2中可以看出,它们发生水交换反应的活化能垒均远低于单核铝,表明一旦发生二聚,所有位点的键合水分子的活性均会明显增加。同时,轴向水分子能垒低于赤道面水分子~20 kJ·mol-1,表明轴向水分子位点是该水合形态发生各种化学反应的活性位点,这一结果也与上述键长变化趋势一致。

表2 各铝形态键合水反应离去反应活化能垒/kJ·mol-1

图3 Al2(μ2-OH)2(H2O)84+水交换反应过程 R1、TS1和I1分别对应于轴向水分子水交换反应的反应物、过渡态和中间体;R2、TS2和I2分别对应于赤道面水分子水交换反应的反应物、过渡态和中间体。
由于二聚铝形态轴向水分子位点活性更高,因此在以下的配体取代影响研究中,以该水分子为研究对象。图4列出了两个轴向水分子分别被OH-和 F-取代时的二聚铝形态 Al2(μ2-OH)2(ƞ-OH)2(H2O)62+和 Al2(μ2-OH)2(ƞ-F)2(H2O)62+,可以发现,取代产物中剩余的两个轴向水分子分别与两个OH-配体和F-配体形成了氢键。通常,由于对位效应的存在[14],阴离子配体的对位配体和中心离子之间的距离与未取代时相比应该会明显增加。从表3可以看出,这一间距从未取代时的1.990 Å增加到2.016 Å,即F-取代时符合对位效应,而在OH-取代中,这一间距并无变化。这是由于在Al2(μ2-OH)2(ƞ-OH)2(H2O)62+中,形成的这一分子内氢键即为金属水合离子中常见的H3O2-结构[15],这一结构通常伴随水分子上的质子向OH基转移,导致这一水分子配体中的氧原子负电荷增加,结果因中心铝离子与水配体之间较强的作用力而抵消了对位效应的影响。

图 4 Al2(μ2-OH)2(ƞ-OH)2(H2O)62+和 Al2(μ2-OH)2(ƞ-F)2(H2O)62+水交换反应过程 R1、TS1和I1分别对应于Al2(μ2-OH)2(ƞ-OH)2(H2O)62+水交换反应的反应物、过渡态和中间体;R2、TS2 和 I2 分别对应于 Al2(μ2-OH)2(ƞ-F)2(H2O)62+水交换反应的反应物、过渡态和中间体;蓝色表示氟离子
对这两个取代形态而言,其水交换反应与未取代的二聚铝形态类似。但是,二者的活化能垒却并不像单核铝一样会随取代而明显下降,而是比较接近于未发生取代的二聚铝形态。其原因在于整个结构由六配位转变为五配位的过程中,由于桥式结构和上述分子内氢键的存在,这种结构刚性导致目标铝离子的六个配体中有三个配体(两个OH桥和一个OH基)难以自由移动以尽可能降低介于六配位和五配位构型之间过渡态的能量,从而抵消了由于团簇电荷下降带来的能垒降低,结果呈现出的能垒与未取代时差别不大,即键合水分子的活性并未随阴离子取代而明显变化。若从单核铝到二聚铝的形成过程看,水解的单核铝与水解的二聚铝活性基本相同,而氟离子取代的单核铝活性略低于氟离子取代的二聚铝活性。
尽管二聚铝形态键合水分子活性研究的实验困难,目前仍无法获得相关实验数据,但是根据上述研究,对二聚铝形态而言,由于羟桥和分子内氢键的存在,其键合水分子活性的变化趋势与单核铝有较大的不同,因此对其它各种聚合铝形态的活性变化研究应针对于具体的构型进行探讨。

表3 二聚铝形态水交换反应过程中键长参数的变化(Å)
3 结论
本文以密度泛函方法研究了从单核铝到二聚铝形态键合水分子的活性变化,同时探讨了OH-和F-两种阴离子配体取代对该活性的影响。结果表明,从单核铝到二聚铝形态键合水分子的活性明显增加,表明聚合促进铝形态参与各种化学反应。OH-和F-配体的引入均导致单核铝形态活性明显增强,而对于二聚铝形态的键合水分子的活性却几乎没有明显影响。表明就单核铝形态而言,pH或F-阴离子配体促进其水解聚合,但单核铝一旦聚合形成二聚铝之后,其促进效果并不明显,换句话说,这种促进效果并非一直存在于整个聚合过程中,这一点在探讨水溶液中铝形态的水解聚合机制时应当予以考虑。此外,这种活性影响结果对于研究典型的环境表界面反应具有一定的意义。
[1]Bogatko S,Geerlings P.Pressure induced speciation changes in the aqueous Al³+system[J].Physical Chemistry Chemical Physics:PCCP,2014,16(39):21383-21390.
[2]Wang W,Liu W,Chang I Y,et al.Electrolytic synthesis of aqueous aluminum nanoclusters and in situ characterization by femtosecond Raman spectroscopy and computations[J].Proceedings of the National Academy of Sciences of the United States of America,2013,110(46):18397-18401.
[3]Loiseau T,Volkringer C,Haouas M A,et al.Crystal chemistry of aluminium carboxylates:From molecular species towards porous infinite three-dimensional networks[J].Comptes Rendus Chimie,2015,18(12):1350-1369.
[4]Ohlin C A,Rustad J R,Casey W H.The energetics of isomerisation in Keggin-series aluminate cations[J].Dalton Transactions,2014,43(39):14533-14536.
[5]Corum K W,Mason S E.Using density functional theory to study shape-reactivity relationships in Keggin Al-nanoclusters[J].Water Research,2016,102(19):413-420.
[6]Casey W H,Rustad J R.Pathways for oxygen-isotope exchange in two model oxide clusters[J].New Journal of Chemistry,2016,40(2):898-905.
[7]Lanzani G,Seitsonen A P,Iannuzzi M,et al.Isomerism of trimeric aluminum complexes in aqueous environments:exploration via DFT-based metadynamics simulation[J].The Journal of Physical Chemistry B,2016,120(45):11800-11809.
[8]Balogh E,Casey W H.High-pressure17O NMR studies on some aqueous polyoxoions in water[J].Progress in Nuclear Magnetic Resonance Spectroscopy,2008,53(4):193-207.
[9]Sun Z,Wang H,Zhang Y,et al.One-dimensional infinite chain structures of[Al2(OH)4(H2O)4]X2(X=I,Br,Cl):an aggregate of Al2species and a precursor of Al(OH)3[J].Dalton Transactions,2013,42(36):12956-12964.
[10]Jin X,Qian Z,Lu B,et al.Density functional theory study on aqueous aluminum-fluoride complexes:exploration of the intrinsic relationship between waterexchange rate constants and structural parameters for monomer aluminum complexes[J].Environmental Science&Technology,2011,45(1):288-293.
[11]Grimme S,Ehrlich S,Goerigk L.Effect of the damping function in dispersion corrected density functional theory[J].Journal of Computational Chemistry,2011,32(7):1456-1465.
[12]Xu X,Zheng J,Truhlar D G.Ultraviolet absorption spectrum of malonaldehyde in water is dominated by solvent-stabilized conformations[J].Journal of the American Chemical Society,2015,137(25):8026-8029.
[13]Nordin J P,Sullivan D J,Phillips B L,et al.An17ONMR study of the exchange of water on AlOH(H2O)52+(aq)[J].Inorganic Chemistry,1998,37(19):4760-4763.
[14]Guégan F,Tognetti V,Joubert L,et al.Towards the first theoretical scale of the trans effect in octahedral complexes[J].Physical Chemistry Chemical Physics 2016,18(2):982-990.
[15]Lang S M,Bernhardt T M,Kiawi D M,et al.Cluster size and composition dependent water deprotonation by free Manganese oxide clusters[J].Physical Chemistry Chemical Physics,2016,18(23):15727-15737.
Investigation on the changes in reactivity of bound water molecules from aqueous monomeric aluminum to dimeric aluminum species
JIN Xiao-yan,LIN Ling,WANG Lin-yu,QIAN Jia-jia,JIANG Xue-yue
(School of Chemistry and Materials Engineering,Fuyang Normal University,Fuyang Anhui236037China)
Density functional theory was employed to investigate the reactivity of bound water molecules on monomeric and dimeric aluminum species in aqueous solution.The computational results suggest that the reactivity of bound water increases from monomeric aluminum to dimeric aluminum species.As for monomeric aluminum,both hydrolysis and the replacement of fluorine ion cause the enhancement of reactivity.However,both hydrolysis and the replacement of fluorine ion has almost no influence on the reactivity of dimeric aluminum species due to the presence of hydroxyl bridge and intramolecular hydrogen bond.
density functional theory;monomeric aluminum;dimeric aluminum;reactivity of bound water molecule
O612.3文献识别码:A
1004-4329(2017)04-029-06
10.14096/j.cnki.cn34-1069/n/1004-4329(2017)04-029-06
2017-07-10
国家自然科学基金项目(21313021);安徽省高校自然科学研究重大项目(KJ2017ZD28);安徽省大学生创新训练计划项目(201610371044)资助。
金晓艳(1977- ),女,博士,副教授,研究方向:环境化学。
猜你喜欢
科教新报(2021年11期)2021-05-12
意林原创版(2017年11期)2017-12-01
四川冶金(2017年6期)2017-09-21
山东工业技术(2016年15期)2016-12-01
首都医科大学学报(2015年4期)2015-12-16
油气地质与采收率(2014年6期)2014-12-16
无机化学学报(2014年12期)2014-02-28
无机化学学报(2014年7期)2014-02-28
无机化学学报(2014年5期)2014-02-28
河南科技(2014年11期)2014-02-27
