
HY分子筛催化异丁烷/丁烯烷基化反应中加成反应的模拟研究
2018-03-05 05:39李永祥代振宇
石油学报(石油加工) 2018年1期
任 奎, 龙 军, 任 强, 李永祥, 代振宇
(中国石化 石油化工科学研究院 催化材料与反应工程国家重点实验室, 北京 100083)
随着我国第六阶段车用汽油标准的实施,汽油中烯烃、芳烃和苯的含量进一步降低[1]。我国汽油池中,催化裂化汽油和催化重整汽油所占比例已超过90%,烷基化和异构化汽油比例不足1%,这导致了我国汽油标准中烯烃和芳烃含量长期高于欧美等发达国家[2-5]。而烷基化汽油以异构烷烃为主(约90%以上),具有辛烷值高和不排放SOx、NOx等优点[6-8]。因此,提高烷基化汽油的比例,是解决我国汽油质量升级的有效途径。
传统的液体酸烷基化技术(硫酸法和氢氟酸法)虽然已工业化多年,却一直存在酸耗高、毒性大等诸多问题[9-11]。而固体酸催化剂,尤其是分子筛催化剂,具有安全、易分离等特点,工业化潜力较大[12]。世界上第一套商业规模的固体酸烷基化装置于2015年8月在中国山东淄博成功启动,且该技术获得了2016年“美国总统绿色化学挑战奖”中的绿色合成路线奖[13]。
异丁烷/丁烯烷基化是一个链式反应,包含了质子化、烯烃加成、氢负离子转移和异构化等反应[6,8]。其中,碳正离子与丁烯异构体之间的加成反应是生产烷基化汽油的主要步骤。在异丁烷/丁烯烷基化体系中所涉及到的所有加成反应中,叔丁基碳正离子与异丁烯和2-丁烯间的加成反应可以直接生成带3个甲基支链的C8碳正离子,这样的碳正离子经过与异丁烷之间的氢负离子转移反应即可生成三甲基戊烷(TMP),这是烷基化反应的理想产物。因此,十分有必要对这两个基元反应的过程进行详细研究。传统的叔丁基碳正离子与异丁烯/2-丁烯的加成反应机理如图1所示。由于在液体酸中,碳正离子是独立存在的,故图1可以用来解释液体酸为催化剂下的加成反应机理。但是,文献[14-16]表明,在固体酸催化剂(如分子筛)催化的反应体系中,碳正离子以“烷氧基团”的构型存在于分子筛孔道内,即中心C原子与分子筛骨架上的O原子以共价键的方式相连,碳正离子不再是独立存在的,而是限定在O原子附近的一定区域内。因此,对于固体酸催化下的烷基化反应,考虑催化剂性质对反应过程的影响,并采用相应固体酸催化剂模型来深入研究反应机理至关重要。
与实验技术相比,分子模拟技术可以从分子和原子尺度,对催化剂和反应物建立模型,并对反应过程的细节进行详细探究。研究表明,在分子筛孔道内,叔丁基碳正离子以叔丁基烷氧基团(t-butylalkoxide, TBA)的形式存在,该结构中的C—O键键长大于标准C—O单键键长[14, 17],故具有一定的反应活性。因此,本研究拟采用分子模拟的方法,以HY分子筛为催化剂模型,对异丁烷/丁烯烷基化反应体系中TBA分别与异丁烯和2-丁烯发生加成反应的过程进行深入探究,并比较二者异同。
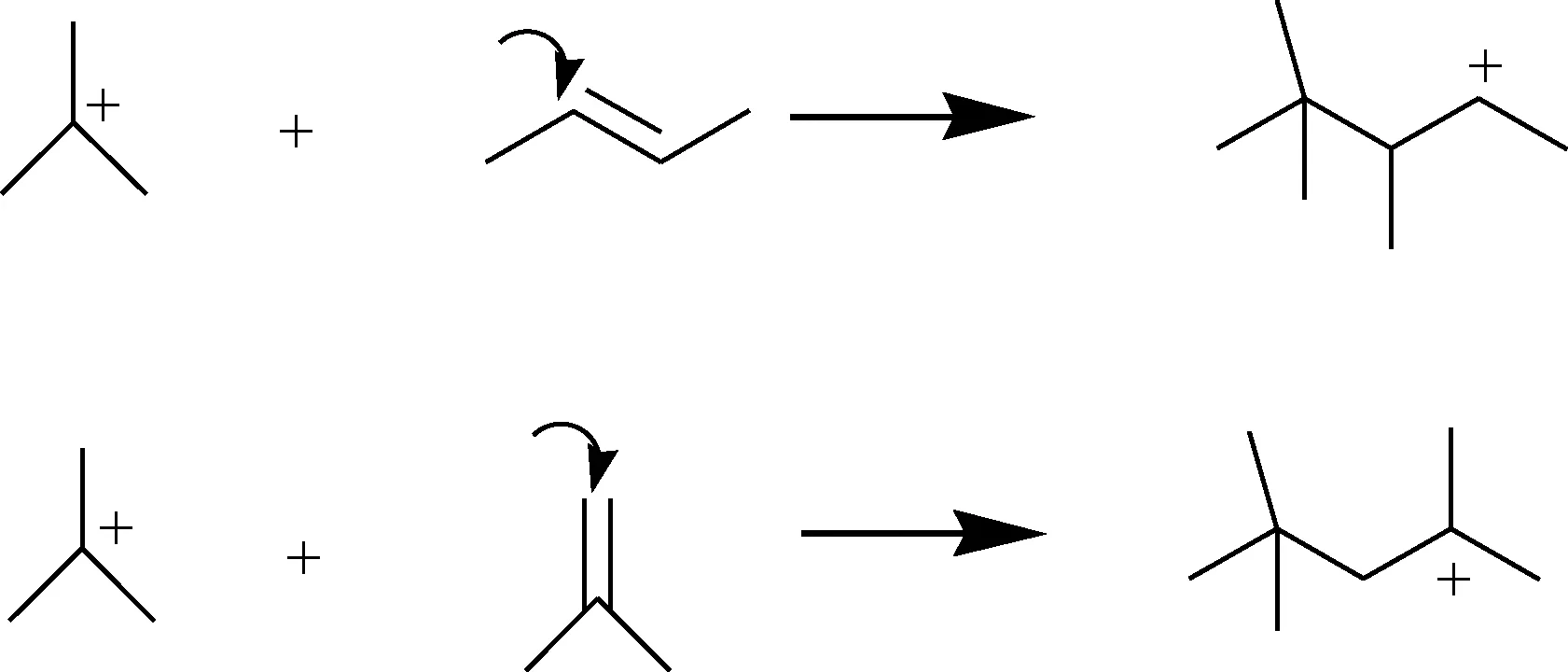
图1 叔丁基碳正离子与异丁烯和2-丁烯加成反应的 反应机理示意图Fig.1 Mechanism schematic of olefin addition reaction of t-butyl carbonium ion and isobutene/2-butene
1 Y分子筛模型的建立和计算方法
1.1 模型的建立
本研究中所用模型为120T HY分子筛模型,该模型截取自FAU分子筛晶体库[18],包含由1个十二元氧环连接的2个超笼,如图2所示。根据理论计算和实验测定,B酸位点的H原子与O1原子相连[19-20]。模型的边界用H原子饱和,形成悬挂的Si—H键。为了保持分子筛的固有特征,所有的Si—H悬挂键与原晶体骨架中的Si—O键的方向一致,Si—H键长固定为0.147 nm。
由于120T的簇模型较大,采用全密度泛函(DFT)方法进行计算非常耗时,而且不切实际。因此,对这样的模型,应该优先考虑采用量子力学与分子力学(Quantum mechanics/molecular mechanics, QM/MM)相结合的方法[21]。该方法将模型继续分成2个部分,内部区域和外部区域。其中,内部区域是反应活性区域,包含分子筛的活性位点和反应物分子;外部区域是分子筛空间环境的代表。B酸位点周围的一些原子和反应物分子为QM区域,其余部分为MM区域。前者以机械嵌入的方式嵌套在分子筛骨架中,采用DFT方法进行计算,所用模块为DMol3[22];后者采用分子力学方法进行计算,所用模块为GULP[23]。为了保证分子筛骨架不发生畸变,模型中所有QM区域原子均处于弛域状态,而所有的非QM区域的原子都固定在其晶体结构位置上。
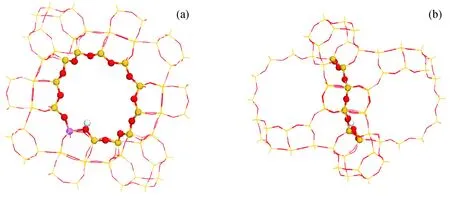
图2 计算所用120T HY分子筛模型的结构示意图Fig.2 Structure schematic of the 120T HY zeolite model used in calculation (a) Front view; (b) Side view
计算所使用的软件为Biovio公司开发的Material Studios 2016。
1.2 计算参数
在DFT计算中,采用Dmol3模块中的密度泛函GGA-PBE方法在DNP基组水平上完成内层活性区域原子和反应物分子的计算。能量、梯度和位移的收敛阈值分别为0.05 kJ/mol、1.05×10-11N/mol和0.0005 nm,所有计算均采用DIIS加速收敛。在QM/MM的计算中,采用UFF力场[24],力场中原子所带电荷采用QEq电荷[25]。采用Additive方式计算能量时,体系总能量ETotal由下式计算[24,26]。
ETotal=EMM+EQM+EQMMM
其中,EQM和EMM分别为QM和MM计算所得到的原子能量,kJ/mol;EQMMM为处于QM和MM边界上的原子对整个体系能量的贡献,kJ/mol。
QMERA模块中过渡态的搜索采用NEB的方法[27]。该方法通过优化2种状态之间的一系列中间产物的构象,可以在已知的反应物和产物之间找到最小能量路径,过渡态位于最低能量路径的鞍点上。
在该方法中,对QM区域进行相关计算前,首先会对QM区域的边界进行氢饱和处理。因此,氢的加入会对活性位点的性质产生影响,即边界效应。为了消除边界效应,笔者采用扩大QM区域的方法来消除[28]。采用22T~36T不同大小的QM区域,对TBA与异丁烯之间加成反应的活化能进行计算,活化能随QM区域大小的变化如图3所示。由图3可以看到,随着QM区域原子的增加,反应能垒大幅度降低。当QM区域扩大到32T时,活化能量值收敛,即此时的QM区域的大小足以消除边界效应,故本研究中QM区域选择32T。

图3 TBA与异丁烯之间加成反应的活化能 随QM区域大小变化的曲线Fig.3 The reaction barrier of addition between TBA and isobutane as a function of QM region
2 结果与讨论
2.1 TBA与异丁烯的加成反应
为了确定TBA与异丁烯的作用位点,采用前线轨道方法对体系进行分析。图4为TBA和异丁烯的前线轨道分布。由图4可以看出,TBA的HOMO主要分布在Al原子所在的六方柱笼内,主要是与Al相邻近的Si、O原子上;LUMO主要分布在TBA的中心C原子和与之相连的O原子上。从图4还可以看出,异丁烯分子的HOMO主要分布在C=C双键以及甲基的C—H原子上,LUMO主要分布在C—C键和C—H键上。TBA叔碳原子的LUMO与异丁烯分子中C=C双键的HOMO对称性匹配;而HOMO轨道电子能量最高,最为活泼,给电子能力强,容易将电子传递到对称性匹配的LUMO轨道,因此,加成反应优先发生在二者之间。
图5为TBA与异丁烯发生加成反应过程各阶段的构象示意图。反应过程中体系内部分原子间的距离和电荷如表1~2所示。

图4 TBA和异丁烯的前线轨道分布Fig.4 The HOMO and LUMO of TBA and isobutene (a) LUMO of TBA (overall structure); (b) LUMO of TBA (local structure); (c) HOMO of TBA (overall structure); (d) HOMO of TBA (local structure); (e) LUMO of isobutene (overall structure); (f) HOMO of isobutene (overall structure)

图5 TBA与异丁烯发生加成反应过程各阶段的构象示意图Fig.5 Structures of various steps during addition reaction of TBA and isobutene (a) TBA+Isobutene (g); (b) TBA+Isobutene*; (c) TS-1; (d) C8 carbonium ion (1); (e) Rotate 180o; (f) C8 alkoxide (1) g——Isolated state; *——Adsorbed state
由图5和表1、表2可以看到,当异丁烯处于游离状态时(见图5(a)),异丁烯分子整体为电中性,即此时异丁烯分子与TBA之间无相互作用。随着异丁烯分子靠近TBA,二者之间会产生较弱的相互作用,形成吸附态的异丁烯分子,其结构如图5(b)所示。此时,异丁烯分子整体所带电荷增加为+0.02 e,说明异丁烯已经向分子筛骨架传递了少量电子。由于二者之间的相互作用,使得C2—O1键长
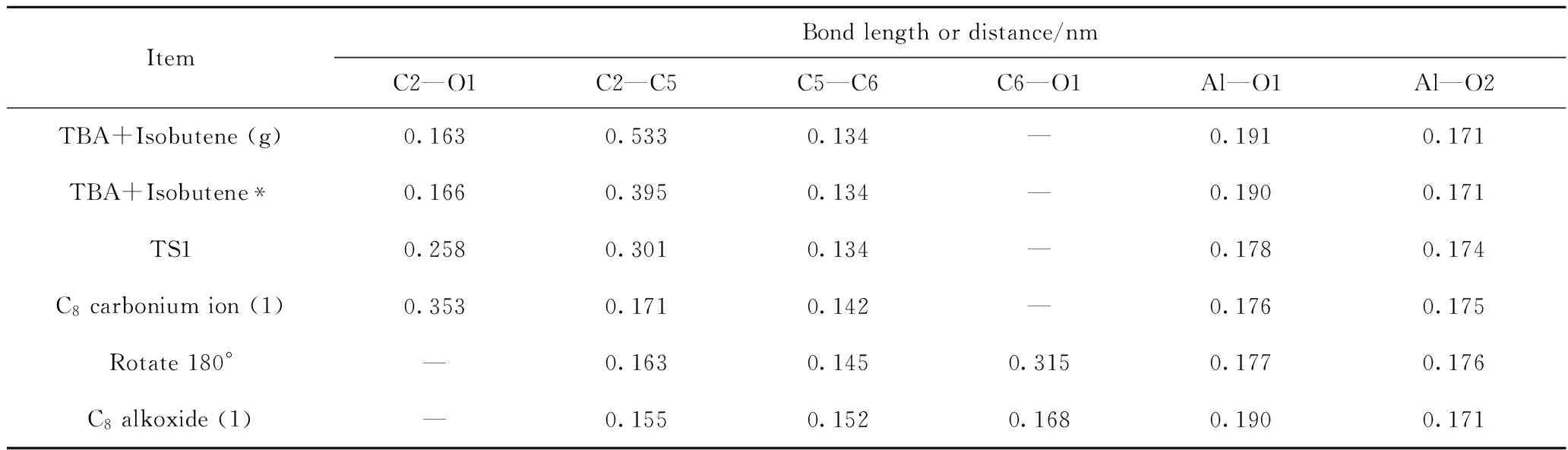
表1 TBA与异丁烯发生加成反应过程中体系内部分原子间距离Table 1 Interatomic distance between atoms during addition reaction of TBA and isobutene
g——Isolated state; *——Adsorbed state
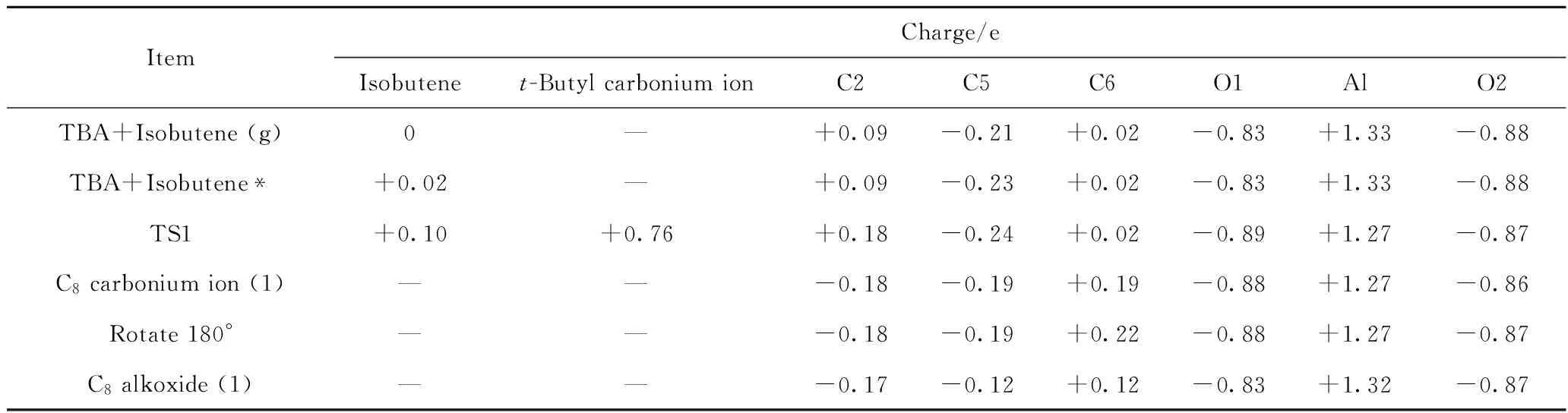
表2 TBA与异丁烯发生加成反应过程中体系内部分原子及原子团的电荷Table 2 Charge of atoms and atomic groups during addition reaction of TBA and isobutene
g——Isolated state; *——Adsorbed state
从0.163 nm增加为0.166 nm,烷氧基团发生反应的活性增加,有远离活性位点的趋势。在该吸附态构象中,C5原子所带电荷为-0.23 e,而C2原子电荷为+0.09 e,二者之间存在较强的静电相互作用。导致C2原子远离O1原子,靠近C5原子,形成吸附态的叔丁基碳正离子过渡态结构(TS1),如图5(c)所示。在该TS1中,C2—O1原子间的距离由0.166 nm增加为0.258 nm,C2—C5原子间距离由0.395 nm减小为0.301 nm。此时,异丁烯分子整体所带电荷从+0.02 e增加为+0.10 e,叔丁基碳正离子整体所带电荷为+0.76 e,说明在这个过程中,异丁烯与TBA之间发生了明显的电子转移,电子从异丁烯C=C双键的HOMO传递到了TBA的C2原子的LUMO。在TS1中,C2、C5原子所带电荷分别为+0.18 e 和-0.24 e,二者之间的静电作用有较大程度的增强,在此作用下,叔丁基碳正离子的中心C原子与异丁烯的C5原子会继续发生电子转移,从而形成C—C单键,这样就生成了如图5(d)所示的C8碳正离子结构,该结构中,C2—C5原子间距离减小为0.171 nm,C2—O1原子间距离增加为0.353 nm,中心C原子由C2转移为C6,C6原子所带电荷为+0.19 e。此后,该C8碳正离子会在活性中心附近以吸附状态的形式发生旋转,并以C6为中心C原子吸附在活性位点O1上(见图5(e)),此时,C6、O1原子所带电荷分别为+0.22 e、-0.88 e,二者之间的静电作用使得C6原子极易与O1原子成键,从而形成如图5(f)所示的C8烷氧基团。
图6为TBA与异丁烯加成反应的能量示意图。由图6可以看到,游离态异丁烯分子进入分子筛孔道形成吸附态异丁烯分子的过程放热为34.6 kJ/mol,克服14.9 kJ/mol的能垒后,形成叔丁基碳正离子的过渡态,过渡态的叔丁基碳正离子与异丁烯形成C8碳正离子,体系能量降低了50.5 kJ/mol,随后C8碳正离子在活性位点旋转180°,并以新的中心C原子吸附在活性氧原子O1上,最终生成C8烷氧基团,从而完成加成反应。
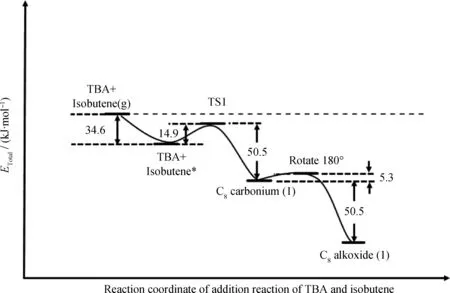
图6 TBA与异丁烯加成反应的能量示意图Fig.6 Energy diagram for addition reaction of TBA and isobutene Energies are given relative to separated 2-butane and TBA. g——Isolated state; *——Adsorbed state
2.2 TBA与2-丁烯之间的加成反应
为了确定TBA与2-丁烯之间发生作用的位点,采用前线轨道方法对体系进行分析,图7为TBA和2-丁烯的前线轨道分布。从图7(a)~(d)可以看出,TBA的HOMO主要分布在Al原子所在的六方柱笼内,主要是与Al相邻近的Si、O原子上;LUMO主要分布在TBA的中心C原子和与之相连的O原子上。从图7(e)~(f)可以看出,2-丁烯的HOMO主要分布在C=C双键以及甲基的C—H原子上,LUMO主要分布在C—H键上。由此可知,TBA的C2原子的LUMO与2-丁烯分子中C=C双键的HOMO对称性匹配;而HOMO轨道电子能量最高,给电子能力强,容易将电子传递到对称性匹配的LUMO轨道,因此,加成反应优先发生在二者之间。
图8为TBA与2-丁烯发生加成反应过程各阶段的构象示意图。反应过程中体系内部分原子间的距离和电荷如表3~4所示。
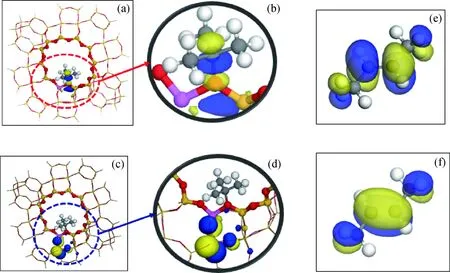
图7 TBA和2-丁烯的前线轨道分布Fig.7 The HOMO and LUMO of TBA and 2-butene (a) LUMO of TBA (overall structure); (b) LUMO of TBA (local structure); (c) HOMO of TBA (overall structure); (d) HOMO of TBA (local structure); (e) LUMO of 2-butene (overall structure); (f) HOMO of 2-butene (overall structure)

图8 TBA与2-丁烯发生加成反应过程各阶段的构象示意图Fig.8 Structures of various steps during addition reaction of TBA and 2-butene (a) TBA+2-Butene (g); (b) TBA+2-Butene*; (c) TS-2; (d) Triangle-structured intermediate; (e) C8 carbonium ion (2); (f) C8 alkoxide (2) g——Isolated state; *——Adsorbed state
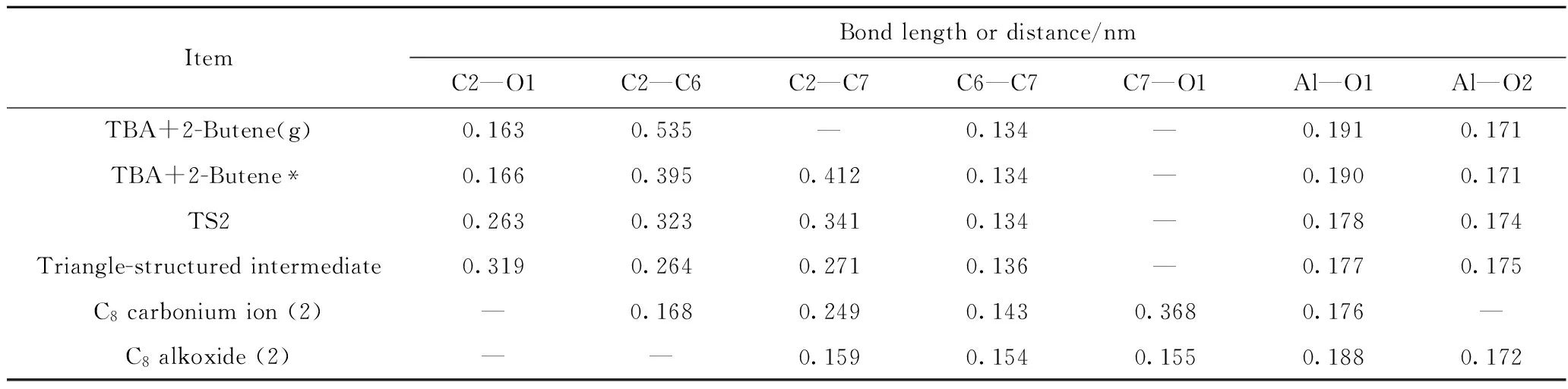
ItemBondlengthordistance/nmC2—O1C2—C6C2—C7C6—C7C7—O1Al—O1Al—O2TBA+2-Butene(g)0.1630.535—0.134—0.1910.171TBA+2-Butene*0.1660.3950.4120.134—0.1900.171TS20.2630.3230.3410.134—0.1780.174Triangle-structuredintermediate0.3190.2640.2710.136—0.1770.175C8carboniumion(2)—0.1680.2490.1430.3680.176—C8alkoxide(2)——0.1590.1540.1550.1880.172
g——Isolated state; *——Adsorbed state
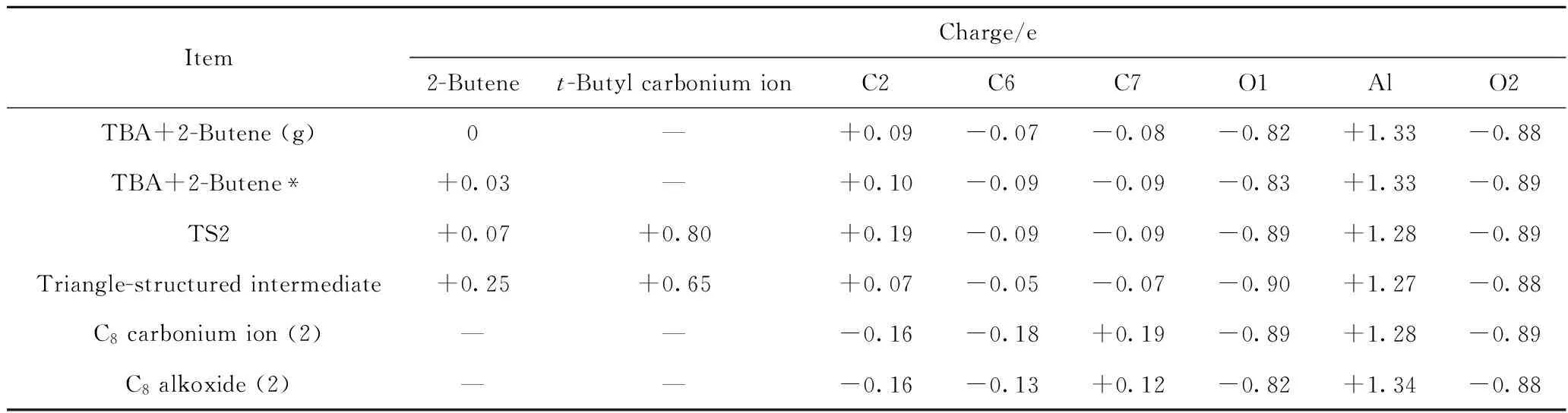
表4 TBA与2-丁烯发生加成反应过程中体系内部分原子及原子团的电荷Table 4 Charge of atoms and atomic groups during addition reaction of TBA and 2-butene
g——Isolated state; *——Adsorbed state
由图8和表3、表4可以看到,当2-丁烯分子处于游离状态时(见图8(a)),2-丁烯分子整体为电中性,即此时2-丁烯分子与TBA之间无相互作用。其中,C6、C7原子所带电荷分别为-0.07 e和-0.08 e,叔丁基烷氧基中心C原子所带电荷为+0.09 e,C2—O1键长为0.163 nm。随着2-丁烯分子靠近TBA,二者之间会产生较弱的相互作用,形成吸附态的2-丁烯分子,其结构如图8(b)所示。此时,2-丁烯分子整体所带电荷为+0.03 e,说明2-丁烯分子已经向分子筛骨架传递了少量的电子。由于二者之间的相互作用,使得C2—O1键的键长从0.163 nm增加为0.166 nm,TBA发生反应的活性增加,有远离活性位点的趋势。在该吸附态构象中,C6、C7原子电荷均为-0.09 e,而C2原子电荷为+0.10 e,C=C双键的C原子与TBA的中心C原子间存在较强的静电作用,使得C2原子逐步远离了O1原子,靠近异丁烯的C=C双键C原子,形成如图8(c)所示的叔丁基碳正离子的过渡态结构(TS2)。此时,C2—O1原子间的距离由0.166 nm增加为0.263 nm,C2—C6、C2—C7原子间距离分别由原来的0.395 nm和0.412 nm变为0.323 nm和0.341 nm。在这个过程中,2-丁烯分子整体所带电荷从+0.03 e增加为+0.07 e,叔丁基碳正离子整体所带电荷为+0.80 e,说明在这个过程中,2-丁烯与TBA之间继续发生电子转移,即2-丁烯C=C 双键的HOMO上的电子传递到了TBA的C2原子的LUMO。在TS2中,C6、C7原子所带电荷均为-0.09 e,而碳正离子中心C原子C2所带电荷增加为+0.19 e,此时,C2与C6、C7之间的作用力增强,会继续靠近,但由于TBA与2-丁烯的加成反应是一个由中心C原子为叔C的结构生成了一个中心C原子为仲C的碳正离子结构,即反应后碳正离子的稳定性降低,因此,叔丁基碳正离子不能与2-丁烯直接生成C8碳正离子,而是首先在C2、C6、C7这3个原子间形成一个“三角形结构”中间产物,如图8(d)所示。在这个中间产物结构中,C2—O1原子间距离增加为0.319 nm,C2与C6、C7原子间距离分别减小为0.264 nm和0.271 nm,C6=C7双键键长由0.134 nm增加为0.136 nm。此时,2-丁烯分子整体电荷从过渡态结构的+0.07 e增加为+0.25 e,碳正离子所带电荷从+0.80 e降低为+0.65 e,说明在形成“三角形结构”中间产物的过程中,2-丁烯分子与碳正离子之间发生了非常明显的电子转移,电子由2-丁烯分子C=C 双键的HOMO传递到TBA中C2原子的LUMO。
要完成加成反应,该“三角形结构”中间产物需要在活性位点旋转180°。该过程中,由于C2原子远离了O1原子,其所带正电荷增加,夺电子的能力增强,容易夺取C6=C7双键的电子,从而形成如图8(e)所示的C8碳正离子(2),并以C7为中心C原子吸附在活性位点O1上。由于该C8碳正离子的中心碳原子C7所带电荷为+0.19 e,与分子筛骨架上带负电荷的O1原子存在较强的静电作用,易形成如图8(f)所示的C8烷氧基团(2),从而完成加成反应。
图9为TBA与2-丁烯加成反应的能量示意图。由图9可以看到,游离态2-丁烯分子进入分子筛孔道形成吸附态2-丁烯分子的过程放热为32.9 kJ/mol,克服29.0 kJ/mol的能垒后,形成叔丁基碳正离子的过渡态;随后过渡态的叔丁基碳正离子与2-丁烯形成“三角形结构”中间产物,体系能量降低了30.5 kJ/mol,“三角形结构”中间产物在活性位点上旋转180°形成C8碳正离子,并以烷氧基团的形式吸附在活性位点O1上从而完成加成反应。
2.3 TBA与异丁烯和2-丁烯发生加成反应的对比
TBA与异丁烯和2-丁烯2个加成反应的异同之处如下:
(1)2个反应的过渡态均为叔丁基碳正离子,但TBA的C—O键断裂后,碳正离子与丁烯之间的作用不同。其中,TS1结构中碳正离子与异丁烯分子整体所带电荷分别为+0.76 e和+0.10 e,TS2结构中碳正离子与2-丁烯分子整体所带电荷分别为+0.80 e 和+0.07 e。可见,在过渡态结构时,异丁烯与碳正离子之间存在更大的电荷转移,即二者间的相互作用力更强。相比之下,2-丁烯与碳正离子之间的电子转移相对明显,即二者间的相互作用相对较弱。因此,异丁烯与TBA发生加成反应时,反应能垒较低,仅为14.9 kJ/mol,小于2-丁烯与TBA发生加成反应时的反应能垒29.0 kJ/mol。
(2)发生反应的丁烯异构体的结构不同导致加成反应路径的本质不同。TBA与异丁烯发生加成反应的反应物和产物的中心C原子类型均为叔C,反应前后中心C原子类型稳定性不变;而TBA与2-丁烯发生加成反应的反应物中心碳原子类型为叔C,产物的中心C原子类型为仲C,中心C原子类型稳定性降低。这就导致了TBA分别与异丁烯和 2-丁烯发生加成反应的路径存在本质的区别。其中,异丁烯可直接与过渡态的碳正离子生成C8碳正离子,而 2-丁烯则首先与过渡态的碳正离子生成“三角形结构”中间产物,再经过旋转生成C8碳正离子,进而完成加成反应。
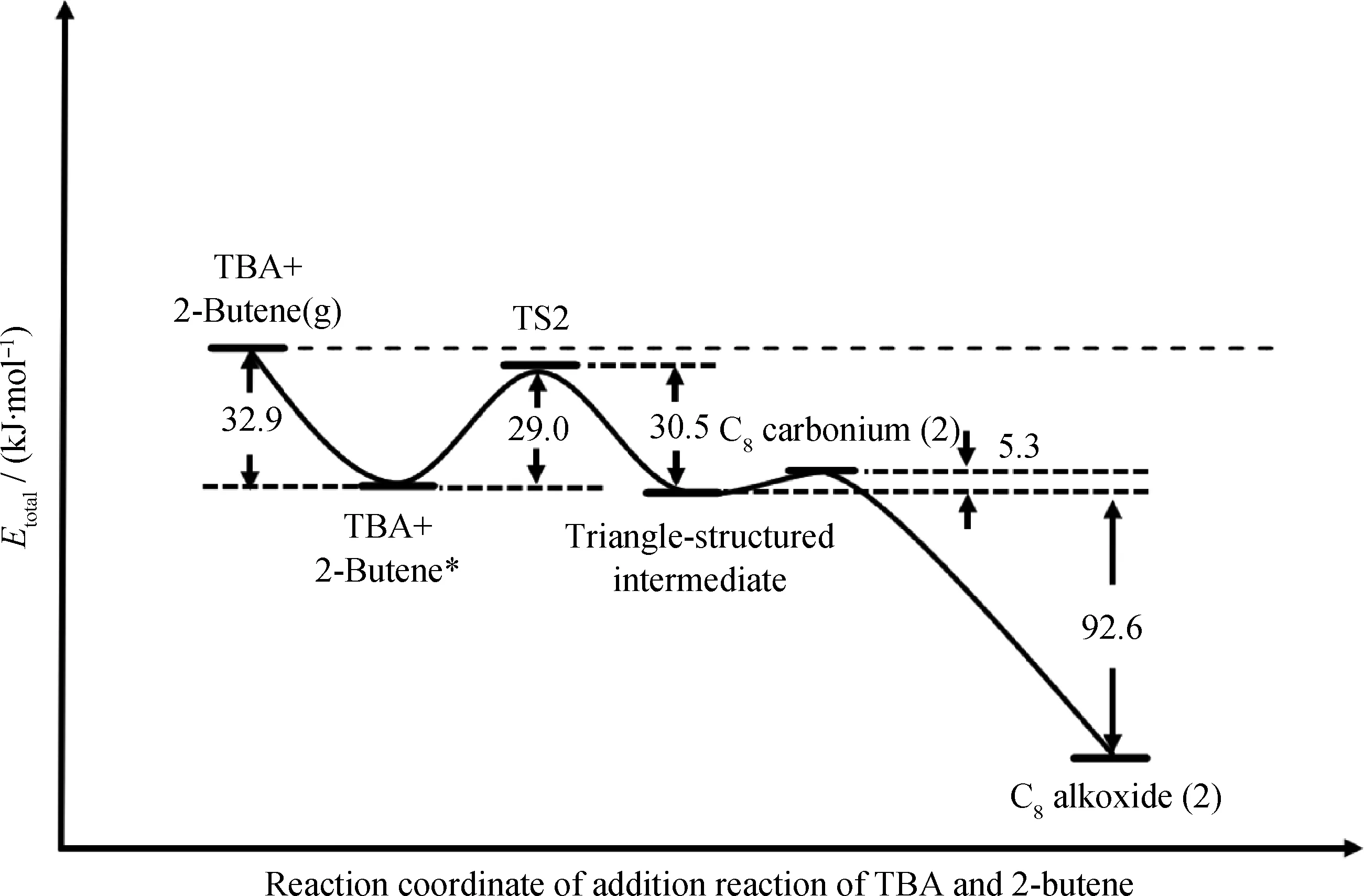
图9 TBA与2-丁烯加成反应的能量示意图Fig.9 Energy diagram for addition reaction of TBA and 2-butene Energies are given relative to separated 2-butane and TBA. g——Isolated state; *——Adsorbed state
3 结 论
(1)TBA与异丁烯分子发生加成反应是通过叔丁基碳正离子过渡态进行的,由于异丁烯分子与碳正离子间的相互作用力较强,故生成该碳正离子需要克服的能垒较低,仅为14.9 kJ/mol。随后异丁烯分子C=C双键中带较多负电荷的端基C原子与碳正离子中带正电的中心C原子形成共价键,生成C8碳正离子,进而形成C8烷氧基团。
(2)TBA与2-丁烯分子发生加成反应也是通过叔丁基碳正离子过渡态进行的,但是与异丁烯相比,2-丁烯分子与碳正离子间的相互作用力相对更弱,故生成该碳正离子需要克服的能垒相对较高,为29.0 kJ/mol。而由于TBA与2-丁烯分子发生加成反应后中心C原子稳定性降低,故需要首先生成“三角形结构”中间产物,随后中间产物在活性位点旋转180°,形成C8碳正离子,最终形成C8烷氧基团完成加成反应。
[1] GB 17930-2016, 中华人民共和国国家标准[S].
[2] 朱玉琴, 司云航, 朱忆宁, 等. 我国车用汽油标准现状及发展趋势[J]. 天然气化工(C1化学与化工), 2014, 39(6): 77-81. (ZHU Yuqin, SI Yunhang, ZHU Yining, et al. Current situation and development trend of motor gasoline standards in China[J]. Natural Gas Chemical Industry, 2014, 39(6): 77-81.)
[3] 王吉云, 张清新. 开好烷基化装置促进汽油质量升级[J]. 石油石化节能与减排, 2012, 2(5): 25-28. (WANG Jiyun, ZHANG Qingxin. Energy gasoline quality upgrade by alkylation operation[J]. Conservation and Emission Reduction in Petroleum and Petrochemical Industry, 2012, 2(5):25-28.)
[4] 许友好, 屈锦华, 杨永坛, 等. MIP系列技术汽油的组成特点及辛烷值分析[J]. 石油炼制与化工, 2009, 40(1): 10-14. (XU Youhao, QU Jinhua, YANG Yongtan, et al. Analysis of octane number and composition characteristics of MIP naphtha[J]. Petroleum Processing Petrochemicals, 2009, 40(1): 10-14.)
[5] 马爱增. 连续重整装置设计参数研究[J]. 石油炼制与化工, 2013,44(6): 64-69. (MA Aizeng. Study of design parameters of CCR reforming units[J]. Petroleum Processing Petrochemicals, 2013,44(6): 64-69.)
[6] 耿英杰. 烷基化生产工艺与技术[M]. 北京: 中国石化出版社, 1993.
[7] 张剑秋. 降低汽油烯烃含量的催化裂化新材料探索[D]. 北京: 石油化工科学研究院, 2001.
[8] 林世雄. 石油炼制工程[M]. 第三版. 北京: 石油工业出版社, 2000: 507-515.
[9] 欧阳健, 郑明光, 张绍良, 等. DUPONT工艺硫酸烷基化装置的腐蚀与防护[J]. 石油化工腐蚀与防护, 2012, 29(6): 31-35. (OUYANG Jian, ZHENG Mingguang, ZHANG Shaoliang, et al. Corrosion of sulfuric acid alkylation unit of DUPONT process and protection[J]. Corrosion & Protection in Petrochemical Industry, 2012, 29(6): 31-35.)
[10] 梁毅. 硫酸法烷基化处理段冲蚀腐蚀分析及处理[J]. 石油化工设备, 2014, 43(5): 104-107. (LIANG Yi. Erosion corrosion analysis and treatment of sulfuric acid alkylation processing section[J]. Petro-chemical Equipment, 2014, 43(5): 104-107.)
[11] LULY M H, POINTNER B E. Hydrogen fluoride compositions: US, 2011152432A1[P]. 2011-06-23.
[12] FELLER A, LERCHER J A. Chemistry and technology of isobutane/alkene alkylation catalyzed by liquid and solid acids [J]. Advances in Catalysis, 2004, 48: 229-295.
[13] Presidential Green Chemistry Challenge: 2016 Greener Synthetic Pathways Award[EB/OL]. https://www.epa.gov/greenchemistry/presidential-green-chemistry-challenge-2016-greener-synthetic-pathways-award.
[14] ROSENBACH N, DOS SANTOS A P A, FRANCO M, et al. Thetert-butyl cation on zeolite Y: A theoretical and experimental study[J]. Chemical Physics Letters, 2010, 485(1-3): 124-128.
[15] MULLEN G M, JANIK M J. Density functional theory study of alkane-alkoxide hydride transfer in zeolites[J]. ACS Catalysis, 2011, 1(2):105-115.
[16] 任奎, 龙军, 任强, 等. HY分子筛催化异丁烷/丁烯烷基化反应中氢负离子转移过程的模拟[J]. 石油学报(石油加工), 2017, 33(6): 703-713. (REN Kui, LONG Jun, REN Qiang, et al. A computational study on the hydride transfer process in butene alkylation with isobutane over HY zeolite[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2017, 33(6): 703-713.)
[17] 鲁玉莹, 李永祥, 龙军, 等. HY催化C4烷基化中异丁烯质子化反应的分子模拟[J]. 石油学报(石油加工), 2014, 30(5): 765-771. (LU Yuying, LI Yongxiang, LONG Jun, et al. Molecular simulation of isobutene protonation in C4alkylation catalyzed by HY zeolite[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2014, 30(5): 765-771.)
[18] OLSON D H, DEMPSEY E. The crystal structure of the zeolite hydrogen[J]. Journal of Catalysis, 1969, 13(2): 221-231.
[19] HILL J, FREEMAN C M, DELLEY B. Bridging hydroxyl groups in faujasite: Periodic vs cluster density functional calculations[J]. The Journal of Physical Chemistry A, 1999,103(19): 3772-3777.
[21] SHERWOOD P, DE VRIES A H, GUEST M F, et al. QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis[J]. Journal of Molecular Structure (THEOCHEM), 2003, 632(1-3): 1-28.
[22] DELLEY B. From molecules to solids with the DMol3approach[J]. The Journal of Chemical Physics, 2000, 113(18): 7756-7764.
[23] GALE J D, ROHL A L. The general utility lattice program (GULP)[J]. Molecular Simulation, 2003, 29(5): 291-341.
[24] RAPPE A K, CASEWIT C J, COLWELL K S, et al. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations[J]. Journal of the American Chemical Society, 1992,114(25): 10024-100035.
[25] RAPPE A K, GODDARD W A. Charge equilibration for molecular dynamics simulations[J]. The Journal of Physical Chemistry, 1991, 95(8): 3358-3363.
[26] BAKOWIES D, THIEL W. Hybrid models for combined quantum mechanical and molecular mechanical approaches[J]. The Journal of Physical Chemistry, 1996, 100(25): 10580-10594.
[27] HENKELMAN G, JNSSON H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points[J]. Journal of Chemical Physics, 2000, 113(22): 9978-9985.
[28] SUN Y, YANG J, ZHAO L, et al. A two-layer ONIOM study on initial reactions of catalytic cracking of 1-butene to produce propene and ethene over HZSM-5 and HFAU zeolites[J]. The Journal of Physical Chemistry C, 2010, 114(13): 5975-5984.
猜你喜欢
——碳正离子的产生及稳定性比较
高中数理化(2022年20期)2022-11-17
石油炼制与化工(2021年2期)2021-02-03
石油炼制与化工(2021年7期)2021-01-14
天津化工(2020年5期)2020-10-15
红领巾·探索(2017年9期)2017-10-26
炼油与化工(2017年4期)2017-09-11
中学生数理化·中考版(2016年10期)2016-12-22
化学教学(2014年1期)2014-02-20
秀·媛尚(2013年6期)2013-09-12
长江大学学报(自科版)(2013年31期)2013-08-11
