
路邓葡萄球菌单宁酶基因的克隆、表达、纯化与改造
2018-05-14 15:33刘鳐胡雪丽钟秋梅等
中国测试 2018年9期
刘鳐 胡雪丽 钟秋梅等


摘要:为提高路邓葡萄球菌(Staphylococcus lugdunensis)单宁酶(Sl-tan)的活性,该文利用化学合成方法获得Sl-tan基因,将该基因连接到重组表达质粒pET43.1-A,再转化到大肠杆菌感受态细胞BL21-DE3中进行表达,通过亲和层析柱纯化,以没食子酸甲酯为底物进行酶活性测定以及酶学性质分析,并基于生物信息学分析,结合定点突变技术对Sl-tan进行人工改造。结果显示,获得的重组单宁酶产量明显增高,最高可达42 mg/L发酵液;酶学性质研究显示该酶在pH 8.0,温度40℃时获得的活性最高(40 U/mg);Ala460突变为Pr0460后的Sl-tan活性可提高82.5%。
关键词:路邓葡萄球菌;单宁酶;三明治结构;定点突变;没食子酸甲酯
中图分类号:TQ925 文献标志码:A 文章编号:1674-5124(2018)09-0063-06
0引言
单宁是一种水溶性多酚化合物,在植物界中广泛存在。单宁中含有丰富的碳源,但由于其含有大量的芳香环类结构,易于螯合金属离子,并与蛋白聚合形成不可溶性沉淀,导致单宁难以被降解利用。一些微生物能够表达单宁酶,将单宁水解为五倍子酸与葡萄糖,为微生物生长提供碳源以及能源物质。单宁酶是已知的唯一能够降解单宁的生物酶类,因此被广泛应用于食品行业、制药行业以及动物饲料的生产中。
目前,单宁酶的生产主要通过产单宁酶菌株的液态深层发酵以及固体发酵两种方法,耗时长、产量低、成本高、难以纯化,且生产的单宁酶通常以粗酶或者菌体形式应用,不利于单宁酶的应用。随着基因工程技术的发展,将单宁酶基因克隆,构建表达质粒,利用表达宿主进行表达的方法也已经取得初步成效。Iwamoto等,首次将乳酸杆菌单宁酶(Lp-tan)基因通过基因扩增构建Lp-tan重组表达质粒,并在大肠杆菌DH5α中成功表达,但是纯化后酶的产量与活性较低。在此基础上,Wu等通过LIC-PCR的方式构建了Lp-tan的重组表达质粒,并优化表达用宿主细胞,最终在大肠杆菌BL21-DE3中实现了Lp-tan的高产量表达,并保持了较高的酶活性。目前为止,已经从米曲霉(Aspergillus oryzae)、乳酸杆菌(Lactobacillus plantarum)以及链霉菌(Streptomyces sviceus)中克隆了單宁酶基因,通过异源重组表达的方法生产单宁酶,取得了较好的结果。
本文通过化学合成获得了路邓葡萄球菌(Staphylococcus lugdunensis)的单宁酶基因Sl-tan,构建原核表达质粒,使其在大肠杆菌宿主中进行表达,并通过与Lp-tan的氨基酸序列比对分析,结合已经报道的Lp-tan的结构(PDB序列号:4JOC)、Lp-tan与底物没食子酸乙酯的结构(PDB序列号:4JOK)、Lp-tan与产物五倍子酸的结构(PDB序列号:4JOH)对Sl-tan进行定点改造。改造后获得的重组Sl-tan活性较改造之前提高了82.5%。
1材料与仪器
1.1材料
大肠杆菌DH5a、BL21-DE3菌株以及质粒pET-43.1-A均为实验室保存;限制性内切酶BamHI和XhoI、T4 DNA连接酶购买自Thermo-Fisher;DNA聚合酶购买自TaKaRa;DNA胶回收试剂盒购买自QIAGEN;定点突变试剂盒购买自北京全式金;商品化米曲霉单宁酶(Wako,Japan);没食子酸甲酯、单宁酸、异丙基硫代半乳糖苷(IPTG)购买自Sigma;咪唑购买自科龙(成都);10KD浓缩管购买自Millipore。
1.2仪器
超净工作台(苏净集团安泰公司SW-CZ-1F);蛋白纯化仪(苏州利穗);全自动高压蒸汽消毒器(上海三申医疗器械有限公司YX280A);摇床(上海智诚);10mLHisTrapHP亲和层析柱(博格隆);超声破碎仪(宁波新芝);高速冷冻离心机(贝克曼,美国)。
2方法
2.1目的基因获取
在NCBI数据库中查找Sl-tan基因(GenBank:KU882097.1),通过化学合成的方法获得完整的基因序列(擎科生物,成都)。
2.2重组载体的构建
本文中利用的重组表达载体为改造后的pET43.1-A(仅保留N端His-tag,并在His-tag后引人烟草花叶病毒酶(TEV)的酶切位点,去除了原先质粒上的S-tag以及NusA-tag),以合成的Sl-tan基因为模板,设计引物序列进行PCR扩增,上游引物序列为CGGATCC-ATGAAA
GACTTTCATATC-ACTCT,下游引物序列为CCTCGAG-CTATTTTTT-ATTAATACTTTCTACC(斜体为BamHI和XhoI的酶切位点及保护碱基),扩增条件为95℃30 s,52℃30 s,72℃2min,35个循环。扩增后的PCR产物与pET43.1-A质粒同时用限制性内切酶BamHI和XhoI进行双酶切,酶切后用胶回收试剂盒纯化。将纯化后的双酶切质粒与PCR产物按照1:3的比例混合后,用T4 DNA连接酶在室温下连接1h,转化DH5a,涂含有卡那霉素的LB平板,37℃培养过夜后,挑取单克隆提质粒,并送公司(擎科生物,成都)测序。
2.3诱导表达与纯化
将测序正确的重组质粒转化表达用大肠杆菌感受态细胞BL21-DE3,涂含有卡那霉素的LB平板,37℃培养过夜后挑取单克隆接种于含有卡那霉素的LB液体培养基中,于37℃,220 r/min培养至OD600值为0.6左右时,加入终浓度为0.5 mmol/L的IPTG继续诱导培养4 h,6000 r/min离心30 min,弃上清,收集菌体。
将收集的菌体用平衡缓冲液(20 mmol/L Tris-HCl,100 mmol/LNaCl,20 mmol/L咪唑,pH 8.0)重悬,超声破碎15 min,离心后收集上清并过5μm孔径滤膜,收集上清并用蛋白纯化仪过10 mL HisTrapHP亲和层析柱,用洗脱缓冲液(20 mmol/L Tris-HCl+100 mmol/L NaCl+300 mmol/L咪唑,pH8.0)梯度洗脱。
2.4 TEV酶切去除N末端组氨酸标签
载体pET43.1-A自身带有18个氨基酸的组氨酸标签,在组氨酸标签后带有烟草花叶病毒酶(TEV)的酶切位点。为了去除组氨酸标签对Sl-tan活性的影响,用TEV酶切去除组氨酸标签。将亲和层析后的Sl-tan,用透析缓冲液(20 mmol/L Tris-HCl+100 mmol/L NaCl,pH 8.0)透析,去除咪唑,然后与TEV按照100:1的比例混合,4℃条件酶切过夜。经酶切后的Sl-tan(加入20mmol/L咪唑)再次过组氨酸亲和层析柱纯化,收集穿透峰样品。
2.5酶活性的测定
酶活性单位U的定义:在温度为40℃,pH为8.0的条件下,每分钟内水解底物生成1μmol产物五倍子酸所需要的酶量,定义为一个酶活性单位U。
单宁酶可以水解没食子酸甲酯以及单宁酸,释放出五倍子酸。通过绕单宁和五倍子酸的特异反应,可以测定单宁酶的活性。以25 mmol/L没食子酸甲酯作为底物,单宁酶可以水解没食子酸甲酯(MG),生成五倍子酸,五倍子酸(GA)可以与绕单宁反应,用NaOH终止反应,测520 nm的吸光度,显色强度与五倍子酸的量成正比。在700μL的反应缓冲液(20 mmol/L Tris-HCl+100 mmol/L NaCl,pH 8.0)中加入0.1μg的单宁酶与40μL摩尔浓度为25 mmol/L的没食子酸甲酯在40℃条件下温育5 min,然后加入150μL质量浓度为0.667%的绕单宁溶液再次在40℃下温育5mm,随后加入100μL摩尔浓度为500 mmol/L的NaOH溶液反应5 min终止反应,用分光光度计检测酶解液在520 nm下的吸光度值。
标准曲线制备:以0.125~1 mmol/L的五倍子酸系列标准溶液与绕单宁反应,测520nm的吸光度,以五倍子酸的浓度为横坐标对纵坐标吸光度值作出标准曲线。
2.6温度与pH对酶活性的影响
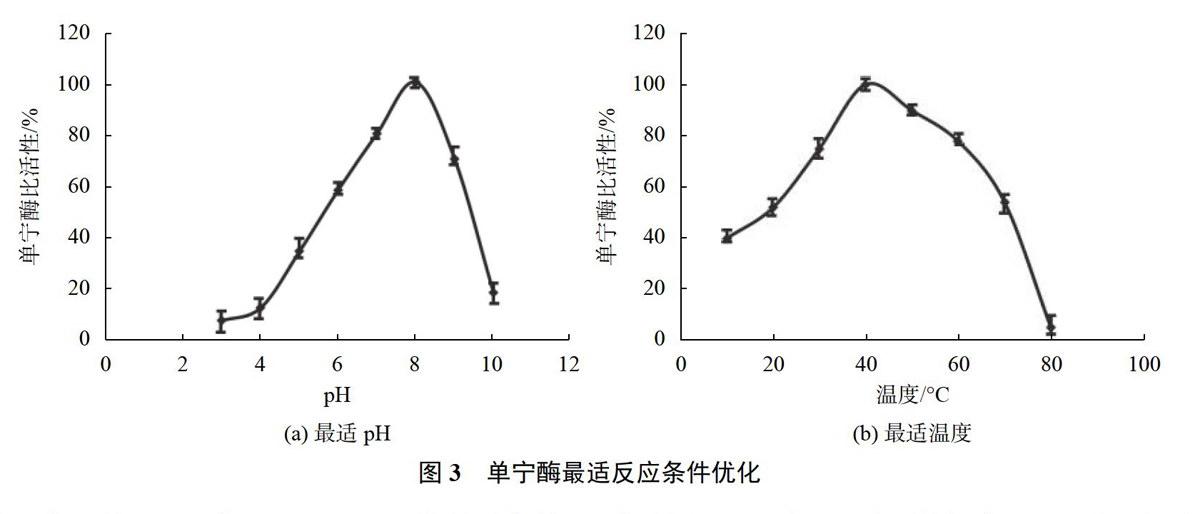
为了研究酶的最适反应温度与pH,将纯化的单宁酶在10~80℃条件下以没食子酸甲酯为底物,在pH值为8.0的条件测定酶的活性,以测得的最高活性值为100%作图。在温度为37℃时,配制pH值为3.0~10.0的缓冲液,以没食子酸甲酯为底物检测酶的活性,以测得最高活性数值为100%作图。
2.7单宁酶的序列分析及定点突变
乳酸杆菌单宁酶具有较高的生物活性,且其三维结构已经被解析,并且根据其结构阐明了单宁酶的水解机制。通过Expasy Align比对分析Lp-tan与Sl-tan单宁酶的差异,并结合已报到的乳酸杆菌单宁酶的结构与水解机制,对差异位点进行定点突变,具体突变方法如下:
通过PCR的方法,以包含Sl-tan基因的质粒为模板,按照定点突变试剂盒试剂盒要求,设计引物(上游引物:TTTAAA CGTAGCCAA-CAG-GAAA-ATGAAGT;下游引物:CTG-TTGGCTACGTTTTAA-ATCATC,斜体表示突变位点),进行PCR扩增(94℃30 s,60℃30 s,72℃8 min,20个循环)。将获得的PCR产物用DpnI酶处理,消化掉模板质粒。将酶切后的PCR产物跑1.0%的琼脂糖凝胶电泳,切胶后回收PCR产物,转化大肠杆菌DH5α,涂平板后于37℃培养24 h后,挑取单克隆于LB培养基中,37℃,200 r/min,过夜培养。提质粒,送测序,将测序结果正确的质粒转化大肠杆菌BL21-DE3感受态细胞,进行诱导,表达、纯化及活性测定,方法同前。
3结果
3.1目的基因PCR扩增及酶切鉴定结果
以合成的Sl-tan基因为模板,进行PCR,1.0%琼脂糖凝胶电泳可见1800 bp左右的DNA片段,与预期一致(见图1)。构建好的重组质粒进行测序,测序结果与NCBI数据库登陆序列一致。
3.2蛋白表达与纯化
用大肠杆菌BL21-DE3作为宿主表达蛋白并于20℃过夜诱导。收菌后,超声破碎,将上清用蛋白纯化仪进行纯化。表达载体pET43.1-A,带有N末端组氨酸标签,表达的目的蛋白前端含有MG-HHHHHHGTENLYFQGS氨基酸序列。破碎后上清过组氨酸亲和层析柱,用高浓度咪唑梯度洗脱,在咪唑浓度为80~170 mmol/L之间出峰。酶切去除N末端组氨酸标签后,再次用镍柱亲和纯化后得到纯度95%以上的单宁酶。10%SDS-PAGE电泳验证(见图2),在分子量67 kD左右有明显的单一目的条带,与Sl-tan中单宁酶分子量大小相符,纯化后,重组单宁酶的产量为42 mg/L菌液。
3.3温度和pH值对酶活性的影响
在不同pH值和不同温度条件下,测定纯化后单宁酶的活性,如图3(a)所示,在pH7~9时,酶可以保持相对较高的活性,在pH值8附近,酶的活性最高;如图3(b)所示,在温度30~60℃时,酶可以保持相对较高的活性,在40℃附近,酶的活性最高。
将纯化后的Sl-tan在40℃,pH 8.0的最适条件下,以没食子酸甲酯为底物进行活性测定,结果显示带有N端组氨酸标签的单宁酶活性为40 U/mg,去除标签后的单宁酶活性并未有明显变化。
3.4 Sl-tan的改造
Sl-tan与Lp-tan的氨基酸序列比对结果显示两者只有21.9%的序列同源性,但在Sl-tan中具有与Lp-tan相同的单宁酶酶活性中心保守序列G-X-S-X-G-G(X代表任意氨基酸,见图4)。在Lp-tan中Pr0356-底物-Ile206形成类似于三明治的结构,Pro356的苯环结构能够稳定底物结合,但是在Sl-tan中,用于形成三明治结构的Pro被Ala取代,从而破坏了三明治结构。通过定点突变的方法在Sl-tan中重建三明治结构(Ala460突变为Pr0460),活性测定结果显示重建三明治结构后的Sl-tan活陛为73 U/mg,與野生Sl-tan相比,酶活性提高82.5%(见表1)。
4讨论
现阶段,我国单宁酶的研究主要集中在产单宁酶的菌株筛选上,对产单宁酶的菌株进行诱变以获取高产菌株。另外,对单宁酶的应用也主要是以粗酶形式,或者直接以发酵后的菌体作为单宁酶使用,由于粗酶或者菌体直接作用可能存在细菌污染,限制了单宁酶的应用。
前期研究中,本课题组通过基因重组表达的方式将乳酸杆菌中的单宁酶基因以及链霉菌中的单宁酶的基因进行克隆,并用大肠杆菌BL21-DE3进行表达,最终获得高产量的单宁酶,并且保持了较高的酶活性。与传统的单宁酶生产方式相比,更易于获得高纯度的单一的单宁酶,有利于单宁酶的工业化应用,但是能够用于重组表达的单宁酶仍然很少。
本文通过基因工程方法将Sl-tan基因进行克隆,并在大肠杆菌BL21-DE3中进行成功表达,获得了高产量的Sl-tan(42 mg/L菌液)。对Sl-tan的活性研究显示,其在pH 8.0,温度40℃的条件下具有最高活性,但进一步的活性测定结果显示其活性较低(40 U/mg)。在之前的研究中,我们报道了Lp-tan的晶体结构,并对单宁酶的水解机制进行了解析。通过对Sl-tan与Lp-tan的氨基酸序列比对发现,两种单宁酶的序列相似度只有21.9%,但是两种单宁酶具有相同的活性中心序列,进一步比对发现,在Lp-tan中形成的有利于底物结合的Pro356-底物-Ile206三明治的结构,在Sl-tan中被Ala460-底物-Ile271所替代(见图4)。因此,本文利用定点突变技术在Sl-tan中将Ala460突变为Pro460,重建三明治结构,活性测定结果显示突变后的Sl-tan活性提高了82.5%,研究结果也进一步说明了在单宁酶中形成的三明治结构有利于底物没食子酸甲酯的结合。
5结束语
本研究通过化学合成的方法获得了Sl-tan的基因,并构建原核表达质粒使其在大肠杆菌中表达,获得了高产量、高纯度的Sl-tan。对Sl-tan定点突变(Ala460突变为Pro460)重建三明治结构后,使Sl-tan的活性提高了82.5%,使其能够更好地应用于工业化生产实际。
