
H2O和CH4在煤表面竞争吸附机理
2018-12-12 09:00林柏泉张祥良
西安科技大学学报 2018年6期
林柏泉,李 永,杨 凯,孔 佳,张祥良
(1.中国矿业大学 煤矿瓦斯与火灾防治教育部重点实验室,江苏 徐州 221116;2.中国矿业大学 安全工程学院,江苏 徐州 221116)
0 引 言
煤与瓦斯突出是指煤矿地下采掘过程中,短时间内从煤壁内部向采掘工作空间突然喷出煤和瓦斯的现象。它能破坏井巷设施、造成人员窒息,是煤矿最严重的灾害之一[1-2]。近年来,许多科研工作者致力于煤层增渗,提高瓦斯抽采效率以减少瓦斯突出[3-4]。生产实践表明,煤层注水可以减少瓦斯涌出量,缓解煤层瓦斯突出和粉尘[5-6]。而热蒸汽驱替作为一种新的增产煤层气方法正被很多学者研究,在煤层注水和热蒸汽驱替过程中,不仅存在煤与H2O,CH4的单独吸附,同时也存在CH4和H2O的共存吸附,因此对比研究煤表面与CH4,H2O的相互作用和CH4,H2O在煤表面的竞争吸附情况,可以为防治煤与瓦斯突出基础研究提供理论依据,也能对提高瓦斯抽采效率及促进煤层气开采提供一定的指导意义。
现有资料表明,成煤过程中,煤本身就含有约5%~16%的水分,水分的存在会对瓦斯的赋存和运移产生较大的影响[7]。冯增朝等认为水分和甲烷同时存在时,水分先于甲烷吸附于煤表面,煤表面吸附位减小,引起甲烷吸附量减少[8]。李晓华等分析了含水量为0%~15%的新景矿3号煤瓦斯解吸速率,认为煤样水分含量与瓦斯解吸速率成负相关,但有临界值[9]。Stuart等选用澳大利亚和中国的煤样通过实验发现水分对高阶煤的影响程度低于低阶煤,含水量增加,吸附热降低,水的存在影响煤对甲烷的吸附量[10]。随着量子化学理论和相关分子模拟软件的成熟,利用密度泛函理论(DFT)研究小分子在煤表面上的作用机理越来越多。相建华等利用蒙特卡洛(GCMC)和分子动力学(MD)模拟研究了兖州煤模型与CH4,CO2,H2O 之间的相互作用,并利用等量吸附热及能量变化数据揭示了3种气体的不同吸附机理[11]。夏阳超等运用密度泛函理论研究了褐煤表面含氧官能团对水分子的吸附机理,得出在不同含氧官能团吸附位点有不同的吸附能[12]。张俊芳等通过分子模拟研究了干燥煤与湿煤在不同温度下的吸附等温线与等量吸附热[13]。王宝俊等选用不同成熟度的5种煤表面结构模型,从分子水平描述了CO,O2,H2O(g),CO2,CH4和H2等6种气体在煤表面的吸附作用,得到了气体吸附作用强弱次序为:CO和O2最强,H2O和CO2次之,CH4和H2最弱[14]。何旭等通过DFT理论论证了在碳模型表面CO2能促进甲烷的脱附[15]。付雨桐等通过DFT理论论证了低阶煤镜质组表面CO2能促进甲烷的脱附[16]。周亚楠等比较了甲烷和水在不同成熟度的煤表面的吸附能,得出各煤表面对水的吸附能大于对甲烷的吸附能。总之,在分子水平,水分子和甲烷分子竞争吸附还未进行深入研究[17]。
煤大分子结构虽然复杂,但很多研究表明煤是由大小不等的石墨片晶或者芳核组成,为了简化模型结构,采用储伟等简化的团簇模型C30H14(9个苯环)作为煤局部表面进行DFT计算[15],比较了水分子与甲烷分子的不同吸附构型的吸附能,吸附平衡距离,分析了甲烷分子和水分子在煤表面吸附时的互相影响情况,从能量自发作用角度分析了水分子和甲烷分子以不同状态存在于煤表面时的能量高低情况,从微观上解释了水分子和甲烷分子的竞争吸附机理。
1 模型与计算方法
煤表面吸附能计算基于Materials Studio 软件包Dmol3模块,因为局部密度泛函计算会增加弱相互作用能,使用色散校正的密度泛函方法[18-19]可以提高计算的准确性。Sony等采用GGA/PBE/Grimme得到了较为精确的值[20],所以文中选择广义梯度近似法(GGA),电子交换关联势采用基于广义梯度近似泛函(GGA)的Perdew-Burke-Ernzerhof(PBE)泛函,计算选取双数值d轨道极化基组(DNP),未设置电子自旋,自洽场的收敛阀值采用10-6,在计算中不考虑基组重叠误差,自洽过程以体系的能量和电荷密度分布是否收敛为依据,精度均优于10-5,位移收敛标准为0.005,力的收敛标准为0.002,能量收敛标准为2×10-5.
C30H14可以认为单层六元环团簇,坐标方向为x×y,即在xy平面内的平面模型。模型首先通过clean工具大致调整结构的不合理部分,然后经过了focite模块和Dmol3模块的Geometry Optimization,分别从力场和密度泛函方法进行构型优化以使得模型能量最小化。
吸附能的计算公式为
EBE=EM+molecule-EM-Emolecule
(1)
式中EM+molecule为煤表面与气体分子的吸附达到平衡态的吸附能;EM为煤表面吸附前的能量;Emolecule为气体分子吸附前的能量。
当2个分子在煤模型表面吸附时,A分子的吸附能公式如下
EABE=EM+A+B-EM+B-Emolecule
(2)
式中EM+A+B为A,B分子在煤表面吸附达到平衡态的吸附能;EM+B为B分子煤表面吸附后的能量。
吸附行为是放热反应,所以吸附能是负值,绝对值越大代表吸附能力越强。图1为分子结构示意图。

图1 分子结构示意图Fig.1 Molecule structure diagram
2 结果与讨论
2.1 甲烷分子和水分子在煤表面的吸附特性
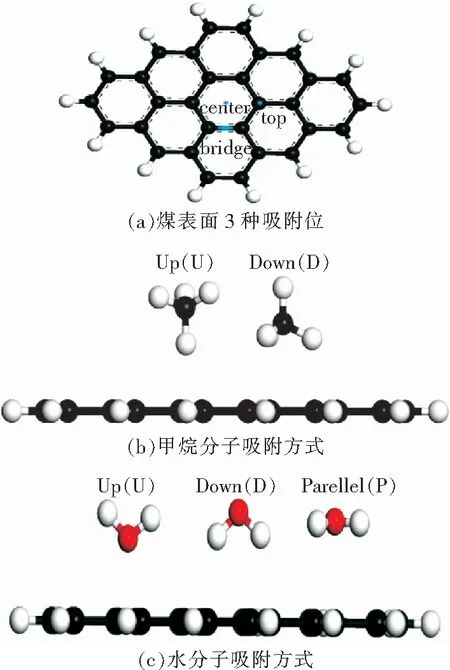
图2 吸附位与吸附方式Fig.2 Adsorption sites and adsorption ways
如图2所示,煤的模型表面考虑3种吸附位,即碳原子上方(top,T),苯环中心(center,C),碳-碳键上(bridge,B)。甲烷分子在煤表面有2种吸附方式,分别为up型(即3个氢原子指向真空方向),down型(3个氢原子指向模型表面)。水分子的吸附考虑3种方式,即水分子平行于模型表面(p)、2个氢原子指向真空方向(up)以及2个氢原子指向模型表面(down)。甲烷分子的吸附方式和模型吸附位共有6种可能的吸附构型组合,即TU,CU,BU,TD,CD,BD,如图3所示。水分子的吸附方式和模型吸附位共有9种可能的吸附构型组合,分别为TU,CU,BU,TD,BD,CD,TP,CP,BP.如图4所示,无论是甲烷分子还是水分子,在不同的吸附构型下,均会产生不同的吸附平衡距离、吸附能。
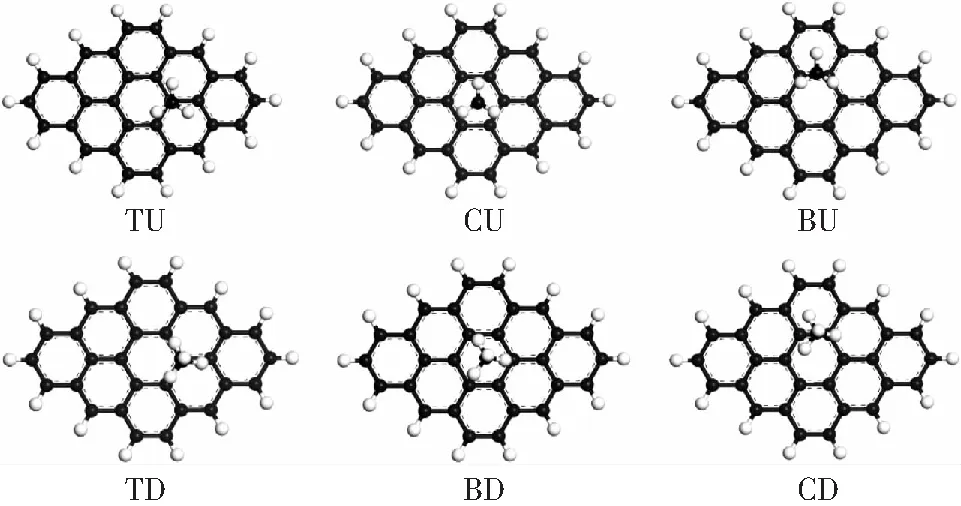
图3 甲烷在煤表面吸附构型Fig.3 Possible configurations of CH4 molecules absorbed onto the coal surface
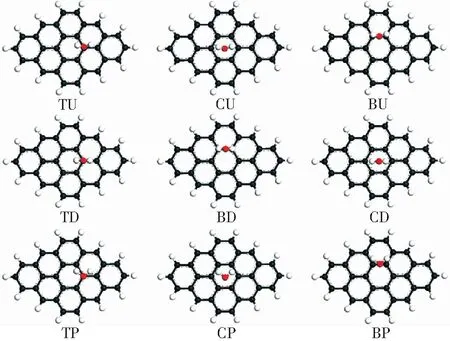
图4 水分子在煤表面吸附构型Fig.4 Possible configurations of H2O molecules absorbed onto the coal surface
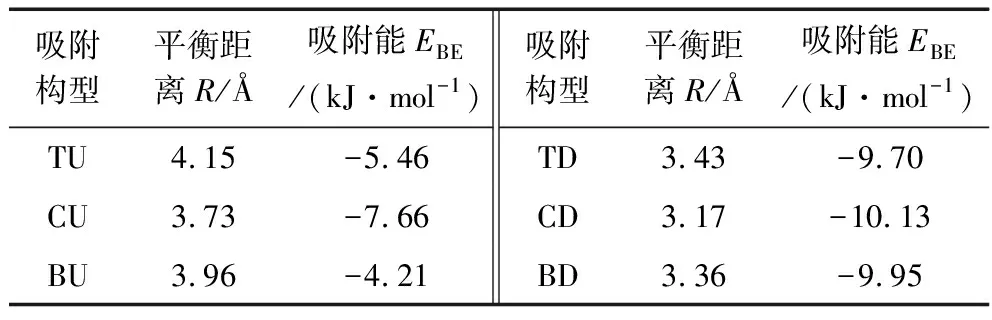
吸附构型平衡距离R/Å吸附能EBE/(kJ·mol-1)吸附构型平衡距离R/Å吸附能EBE/(kJ·mol-1)TU4.15-5.46TD3.43-9.70CU3.73-7.66CD3.17-10.13BU3.96-4.21BD3.36-9.95
由表1可以看出,在甲烷分子的所有吸附构型中,甲烷的吸附能介于-4.21~-10.13 kJ/mol之间,吸附平衡距离介于3.17~4.15 Å之间。
分析吸附方式的影响可以得出,甲烷分子以down型(CD,TD,BD)吸附方式进行吸附时,吸附平衡距离明显小于up型(TU,CU,BU),吸附能明显大于up型。
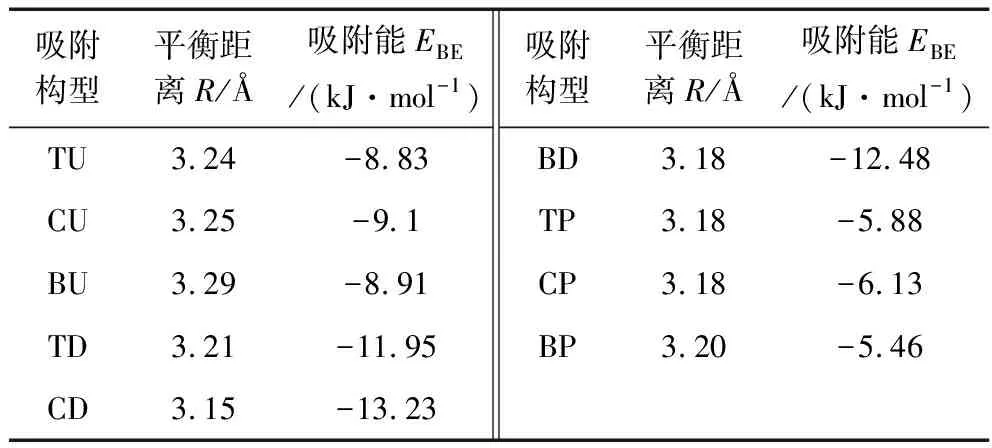
表2 H2O在不同吸附构型下的吸附平衡距离、吸附能Table 2 Equilibrium intermolecular distances(R)and adsorption energies(EBE)of H2O on the surface of coal
分析吸附位的影响,在up型3种不同的吸附位情况下(TU,CU,BU),以CU吸附构型的吸附平衡距离最近且吸附能最大,这说明在up型吸附方式时,以center作为吸附位所构成的CU吸附构型最稳定,这就证明center吸附位要比其它2个吸附位更有利于吸附。
由表2可以看出,在水分子的所有吸附构型中,水的吸附能介于-5.46~-13.23 kJ/mol之间,吸附平衡距离介于3.15~3.29 Å之间。
分析吸附方式的影响可以得出,水分子分子以down型(CD,TD,BD)吸附方式进行吸附时,吸附能与电荷转移量均明显大于up型(TU,CU,BU)与parellel型(TP,CP,BP)。吸附平衡距离变化不大。
分析吸附位的影响,可以看出在up型3种不同的吸附位(TU,CU,BU),down型3种不同的吸附位(TD,CD,BD)和parellel型3种不同的吸附位(TP,CP,BP),CU,CD,CP吸附能,吸附平衡距离均较小,可以得出center位有利于水分子吸附。
从上文分析可知,无论是甲烷分子或是水分子,在煤表面吸附时,相较吸附位,吸附剂的吸附方式对吸附行为的影响较大,且均以CD型吸附构型为最稳定吸附,选取2种分子在最稳定吸附的情况下进行吸附能量对比,如图5所示。通过吸附能对比可以明显看出,甲烷分子在煤表面的吸附能(绝对值)小于水分子在煤表面的吸附能,这就说明水分子比甲烷分子更易吸附在煤表面。
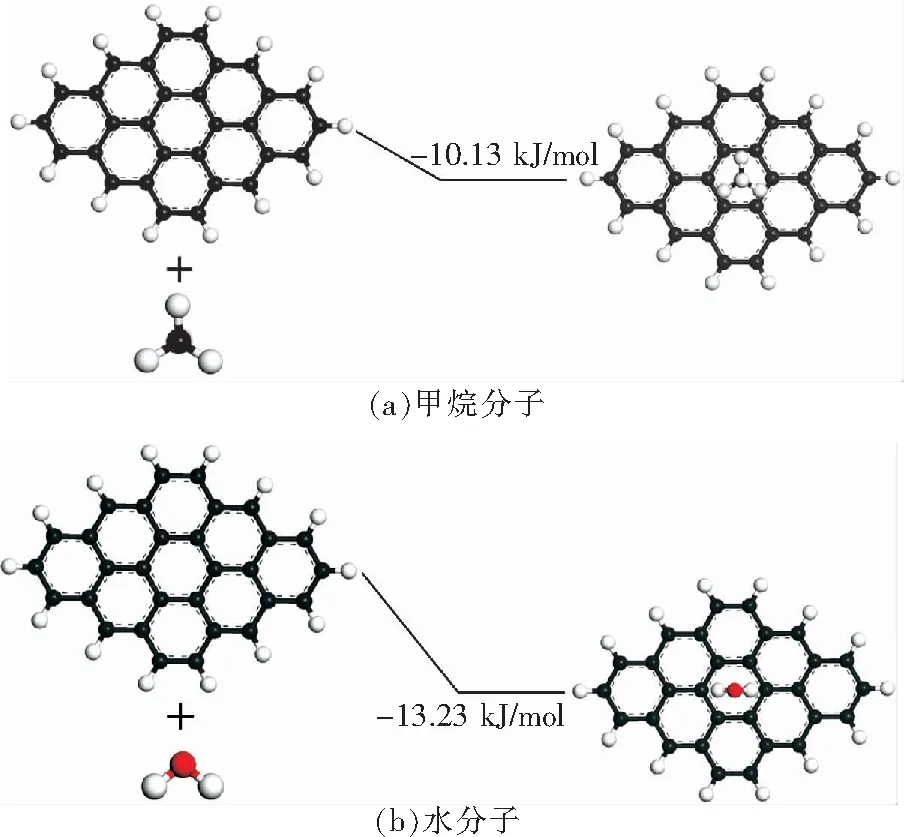
图5 甲烷分子和水分子最稳定吸附构型吸附对比Fig.5 Comparison between CH4 and H2O in the most stable configuration
2.2 水分子和甲烷分子竞争吸附
2.2.1 甲烷分子在已吸附水分子煤表面的吸附特性
将水分子以down型吸附方式、center吸附位、吸附距离为3.21 Å的状态吸附在煤模型上,作为初始吸附模型。在进行甲烷分子吸附时,为甲烷分子选取4个吸附位,如图6(a)所示。其中,甲烷分子在①和③号吸附位所得到的吸附能,与上文中所得到的甲烷分子单独吸附在center位时(无水分子影响)的吸附能相比较,甲烷分子吸附在②和④号位时,与上文中的bridge位相比较。将①②③④4种吸附位状态下甲烷分子和已吸附水分子的距离定义为s1,s2,s3,s4,取s1=1.33 Å,s2=2.67 Å,s3=4.00 Å,s4=5.34 Å,然后计算这4种情况下的吸附能,得到能量-距离图如图7所示。
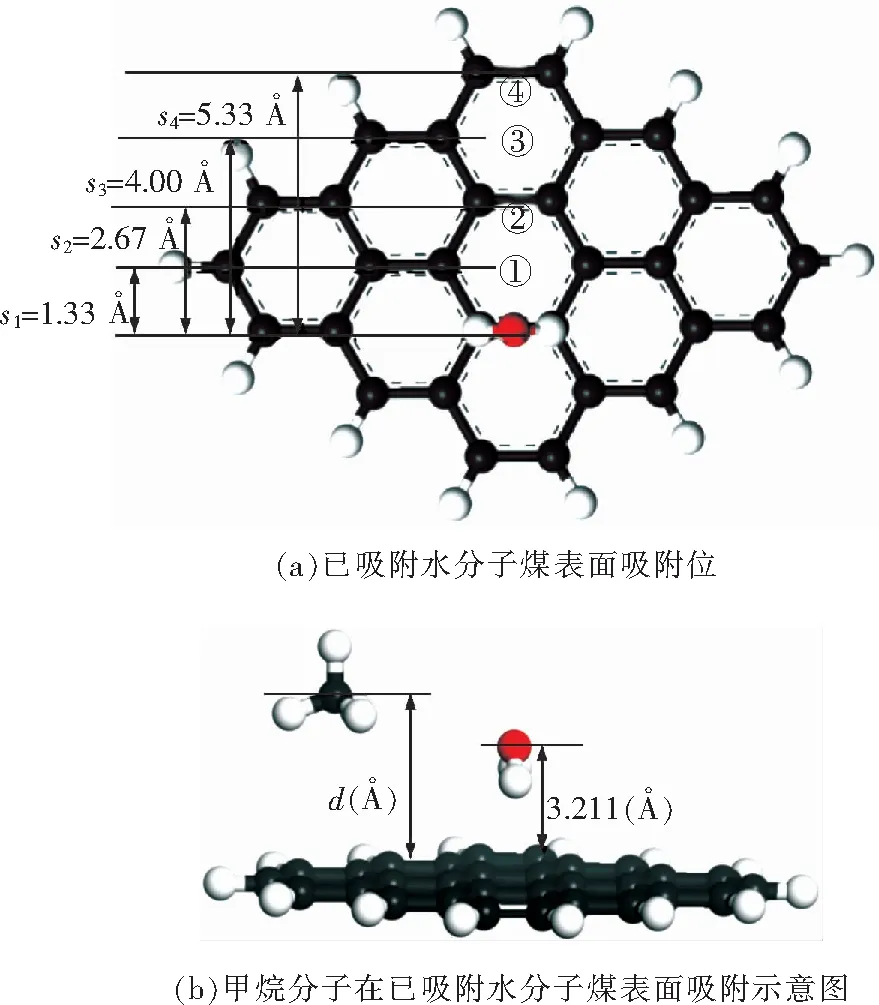
图6 水-煤表面吸附示意图Fig.6 The schematic diagram of adsorption on the H2O-coal surface
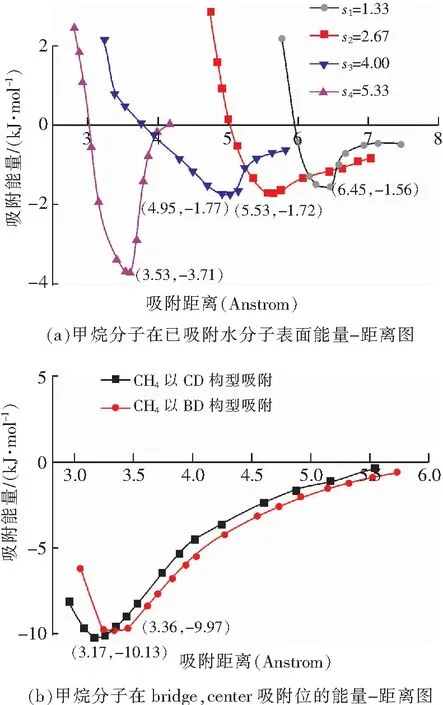
图7 甲烷分子吸附距离-能量图Fig.7 The energy of CH4 molecules with different distances
从图7(a)可以看出,甲烷分子在已吸附水分子的煤表面进行不同距离吸附时,吸附能显著减小。s1=1.33 Å时,含水分子的煤模型甲烷分子吸附能为-1.56 kJ/mol,而甲烷分子吸附在无水分子的煤模型上吸附能为-10.13 kJ/mol,能量释放减少了8.57 kJ/mol,因此更不稳定。同样,在s2=2.67 Å时,能量释放减少8.23 kJ/mol;s3=4.00 Å时,能量释放减少8.8 kJ/mol;s4=5.33 Å时,能量释放减少6.78 kJ/mol.这表明有水分子存在时,甲烷分子吸附放能更少,因此更不稳定。
对此状态下所得到的甲烷分子吸附平衡距离进行分析得:甲烷分子和水分子的距离为s1时,甲烷分子的吸附平衡距离为6.45 Å,而甲烷分子在无水分子影响时的吸附平衡距离为3.17 Å,平衡距离增大了3.28 Å;距离为s2时,平衡距离增大了2.17 Å;距离为s3时,增大了1.78 Å距离为s4时,增大了0.18 Å.这同样表明有水分子存在时,甲烷分子的吸附平衡距离增大,因此更不稳定。
模拟表明,煤表面吸附了水分子后,会影响甲烷分子的吸附行为,水分子的存在使甲烷分子的吸附能显著降低,吸附平衡距离增大,吸附变得不再稳定。通过给定的4个不同距离(s1,s2,s3,s4)所得到的吸附能可以得出,当甲烷分子均受到水分子影响时,距离水分子越远的甲烷分子,受到位阻效应的影响越小,直观表现为吸附能越大、吸附平衡距离越小,即吸附越稳定。因此可以推测当2种分子距离很远时,甲烷分子的吸附能和吸附平衡距离会回归到正常水平。
2.2.2 水分子在已吸附甲烷分子煤表面的吸附特性
将甲烷分子以down型吸附方式、bridge吸附位、吸附距离为3.21 Å的状态吸附在煤模型上,作为初始吸附模型。在进行水分子分子吸附时,为水分子选取4个吸附位,如图8(a)所示。其中,水分子分子在①和③号吸附位所得到的吸附能,与上文中所得到的水分子分子单独吸附在center位时(无水分子影响)的吸附能相比较,水分子吸附在②和④号位时,与上文中的bridge位相比较。将①②③④4种吸附位状态下甲烷分子和已吸附水分子的距离定义为s1,s2,s3,s4,取s1=1.33 Å,s2=2.67 Å,s3=4.00 Å,s4=5.33 Å,然后计算这4种情况下的吸附能,得到能量-距离图如图9所示。

图8 甲烷-煤表面吸附示意图Fig.8 The schematic diagram of adsorption on the CH4-coal surface
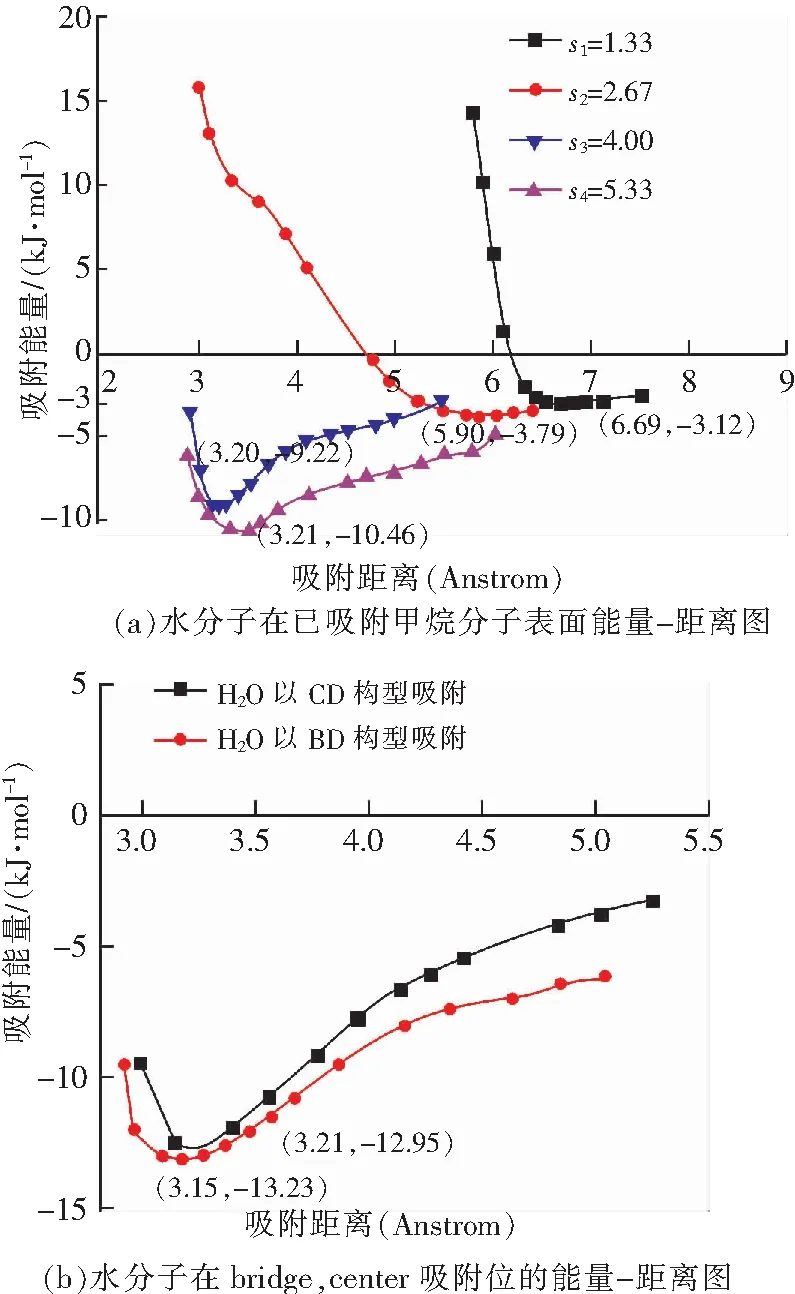
图9 水分子吸附距离-能量图Fig.9 The energy of H2O molecules with different distances
从图9(a)可以看出,水分子在已吸附甲烷分子的煤表面进行不同距离的吸附时,吸附能亦有一定减小,s1=1.33 Å时,含甲烷分子的煤表面水分子吸附能为-3.12 kJ/mol,而水分子吸附在无甲烷分子的煤表面上吸附能为-13.23 kJ/mol,能量释放减少了10.11 kJ/mol,同样,在s2=2.67 Å时,能量释放减少9.25 kJ/mol;s3=4.00 Å时,能量释放减少4.03 kJ/mol;s4=5.33 Å时,能量释放减少2.32 kJ/mol.可以得出水分子的吸附能随着s增大,逐渐增大,趋近于单独吸附时的吸附能。
对此状态下所得到的水分子吸附平衡距离进行分析得:甲烷分子和水分子的距离为s1时,甲烷分子的吸附平衡距离为6.69 Å,而甲烷分子在无水分子影响时的吸附平衡距离为3.15 Å,平衡距离增大了3.54 Å;距离为s2时,平衡距离增大了2.68 Å;距离为s3时,增大了0.05 Å;距离为s4时,恢复到单独吸附时的平衡距离。
对比甲烷在已吸附水分子煤表面的吸附,在两分子距离较小时,(s1=1.33 Å,s2=2.67 Å)甲烷在已吸附水分子表面的吸附能下降幅度小于水分子在已吸附甲烷表面的吸附能下降幅度,吸附平衡距离均增大较多,分析原因可能是因为甲烷分子相较水分子拥有更大的分子直径,更强的位阻效应导致水分子吸附能的下降。在两分子距离较大时(s3=4.00 Å,s4=5.33 Å),甲烷分子在已吸附水分子表面的吸附能下降幅度大于水分子在已吸附甲烷分子表面的吸附能下降幅度,吸附平衡距离后者回归更快,分析原因是因为在较远距离时,水分子对甲烷分子的吸附影响作用要远大于甲烷分子对水分子吸附的影响。而且水分子对甲烷分子吸附行为的影响范围也应大于甲烷分子对水分子吸附的影响范围。间接表明在水分子与甲烷分子共存时,会发生竞争吸附现象,且水分子的吸附处于优势地位,能占据有利吸附位,并影响周围甲烷分子的吸附,促进甲烷分子的脱附,进而增加甲烷产量。
2.3 水分子吸附促进甲烷分子脱附的理论分析
为了更好解释甲烷分子和水分子在煤模型表面的竞争吸附行为,提出以下4种反应作进一步论证

(1)
(2)
(3)
(4)
由图7和图9可以看出,吸附能在大于吸附平衡距离之后,会逐渐减小直至趋于稳定。当垂直距离足够大时,就可以认为吸附剂处于脱附状态。因此,选取距煤模型表面垂直距离为12.00 Å认为吸附剂处于脱附状态,用g表示;选取垂直距离分别为3.17与3.15 Å(即甲烷分子和水分子的平衡距离)认为甲烷分子和水分子均处于吸附状态,用s表示。以反应(1)为例,初始状态表示水分子和甲烷分子均处于脱附状态,最终状态表示甲烷分子处于吸附状态,水仍处于脱附状态,初始状态和最终状态下吸附质和吸附剂的总能量用E1和E2表示,定义ΔE1=E2-E1,当ΔE1<0表示反应可自发,绝对值越大表示反应越容易发生。反应(1)至(4)的总能量如图10所示。
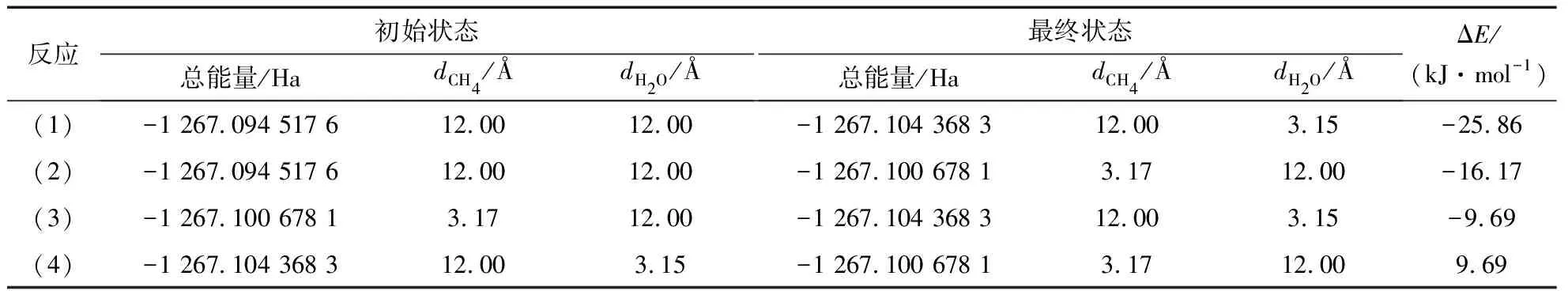
表3 4种不同反应下的初始状态与最终状态Table 3 Initial state and finial state of four reactions
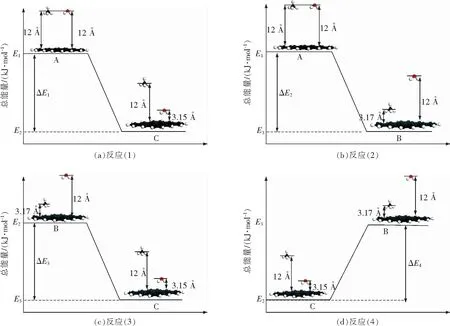
图10 竞争吸附过程中的反应Fig.10 Different reaction of the comparison adsorption process
由图10可得,反应(1),(2),(3)的ΔE均小于0,反应(4)ΔE大于0,为反应(3)的逆反应。这表明,反应(1),(2),(3)这3种吸附反应是自发的,反应(4)的ΔE大于0,表明反应不能自发,一定程度上说明CH4不能促使H2O脱附。还可以看出,在4种反应中,反应(3)最终的能量E3为最低,表示它为最稳定的吸附状态;ΔE1>ΔE2>ΔE3,表明反应(1)最易发生,反应(3)与(4)是初始最终状态相逆的反应,反应(3)的自发性代表水分子的吸附能促进甲烷分子的脱附,反应(4)表示甲烷分子不能促进水分子的脱附,同时也表明在煤中,水分子比甲烷分子有更强的吸附能力。
3 结 论
1)在煤局部表面,得出水分子和甲烷分子均以down型吸附方式,center位吸附时吸附最稳定,两种分子以CD吸附构型进行吸附时,水分子与甲烷分子的吸附能分别为-13.25,-10.13 kJ/mol,水分子比甲烷吸附更稳定;
2)研究了甲烷分子在已吸附水分子煤表面的吸附行为,数据表明在距离水分子不同距离进行吸附,吸附能均显著减小,吸附平衡距离明显增大,一定程度表明水分子的吸附促进甲烷的脱附,对比水分子在已吸附甲烷分子煤表面的吸附,发现水对甲烷吸附能力的影响大于甲烷对水吸附能力的影响;
3)从能量自发角度研究了水分子与甲烷分子共存时的竞争吸附行为,研究发现水分子处于吸附态,甲烷分子处于脱附状态能量最低,最稳定。其他状态易自发到此状态,表明在水与甲烷共存时,煤优先吸附水,使甲烷吸附到不稳定的吸附位,容易脱附。
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
科教新报(2021年11期)2021-05-12
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
安徽农业科学(2018年1期)2018-05-14
意林原创版(2017年11期)2017-12-01
北京航空航天大学学报(2017年10期)2017-04-20
火炸药学报(2015年2期)2015-03-07
基层建设(2014年12期)2014-10-21
航天返回与遥感(2014年4期)2014-07-31
