
简化晶种导向液法合成低硅铝比SAPO-34分子筛
2019-01-24 03:25马晓月陈艳红
中国石油大学学报(自然科学版) 2019年1期
张 强, 马晓月, 刘 璐, 陈艳红
(1.中国石油大学(华东)重质油国家重点实验室,山东青岛 266580; 2.东营市海科新源化工有限责任公司,山东东营 257000; 3.中国石油大学胜利学院化学工程学院,山东东营 257061)
SAPO-34分子筛因其独特的三维交叉通道和八元环通道菱沸石(CHA)笼状结构,在甲醇制低碳烯烃工艺(MTO)中表现出优良的催化性能[1-4]。低硅铝比SAPO-34具有较多的弱酸中心,可以减缓积炭中多甲苯向多环芳烃的转化,增强分子筛的抗积碳能力,并提高低碳烯烃选择性[5-7]。低硅铝比SAPO-34分子筛常采用水热晶化法制备,合成中一般需要加入价格昂贵的四乙基氢氧化铵为模板剂[8]或昂贵的模板剂与昂贵的硅源或铝源配合使用[9]。由于初始凝胶体系中硅浓度较低,致使硅进入骨架的驱动力较小,合成的低硅SAPO-34分子筛结晶度较低,并且很难得到纯的晶相[10]。改进措施包括超声波[11]或微波辅助晶化[12-13]、添加矿化剂[14]、添加晶种或导向剂等。其中采用超声波或微波辅助晶化的方式过于复杂,限制了在工业中的应用;添加矿化剂的方式多数是引入HF,而HF具有强腐蚀性。因此使用廉价模板剂辅助添加晶种或导向剂的制备方法具有更广阔的工业应用前景。大多数导向剂合成法过程比较复杂,如管洪波[15]等通过向凝胶中添加HF,并先对凝胶进行老化和高温晶化,再与超声法相结合制备SAPO-34,该方法不仅使用了超声波合成方法、还应用了强腐蚀性的HF。狄春雨[16]等以少量四乙基氢氧化铵为模板剂先制备导向剂,然后再水热晶化制备SAPO-34,该方法虽然在一定程度上减少了模板剂的用量,但仍需要使用昂贵的季铵盐模板剂,并且合成的分子筛的硅铝比较高,为0.5。针对低硅铝比SAPO-34(硅铝比低于0.4)合成困难、导向剂法合成过程复杂并需要使用昂贵模板剂及硅铝源的问题,笔者用三乙胺为模板剂,分别以硅溶胶和拟薄水铝石为硅源和铝源、采用简化的晶种导向液法合成硅铝比为0.2的SAPO-34,并与晶种法合成样品进行对比,详细考察两种合成方法对分子筛物性和催化性能的影响。
1 实 验
1.1 分子筛的制备
1.1.1 晶种法
分子筛的合成:将晶种、硅源、铝源、磷源、三乙胺和水混合反应,并剧烈搅拌4 h,形成SAPO-34分子筛的初始凝胶。然后将凝胶装入晶化釜,先低温110 ℃水热晶化4 h,再高温180 ℃水热晶化不同时间;晶化后的产物离心、烘干,焙烧得到分子筛样品。初始凝胶中各组分的物质的量比例为SiO2∶Al2O3∶P2O5∶TEA∶H2O=0.2∶1∶1∶4∶50;晶种为购买商业成品SAPO-34(购自南开催化剂厂),加入量为初始凝胶混合物总质量的1.5%。
1.1.2 晶种导向液法
(1)导向液的制备:将晶种、硅源、铝源、磷源、三乙胺和水混合搅拌,并剧烈搅拌4 h,配置成合成导向液的凝胶。将均匀的凝胶装入晶化釜,180 ℃水热晶化1 h,取出后得到乳白色均匀透亮的凝胶溶液,以此溶液作为晶种导向液。晶种导向液凝胶中各组分的物质的量比例为SiO2∶Al2O3∶P2O5∶TEA∶H2O=0.2∶1∶1∶4∶50,晶种为购买商业成品SAPO-34(购自南开催化剂厂),晶种加入量为初始凝胶混合物总质量的1.5%。
(2)分子筛的合成:将硅源、铝源、磷源、三乙胺和水混合反应,并剧烈搅拌4 h,加入步骤(1)中所得到的晶种导向液,并剧烈搅拌4 h,形成SAPO-34分子筛的初始凝胶,装入晶化釜,先低温110 ℃水热晶化4 h,再高温180 ℃水热晶化不同时间。水热晶化后的产物收集、离心、烘干,焙烧得到分子筛样品。初始凝胶中各组分的物质的量比例为SiO2∶Al2O3∶P2O5∶TEA∶H2O=0.2∶1∶1∶4∶50。晶种导向液的用量为凝胶用量的1/22。
1.2 分子筛的表征
采用荷兰帕纳科公司的X’ Pert PRO MPD衍射仪进行分子筛X-射线(XRD)表征,Cu靶Kα射线,X-射线波长为0.154 874 nm,管电压为40 kV,管电流为40 mA,扫描速度为 10°/min,扫描范围为2θ=5°~60°。采用美国Termo NicoletCo.公司生产的NexusTM型傅里叶变换红外光谱仪(FT-IR)进行分子筛骨架振动测试,以DTGS检测器,采集次数为32,分辨率为4,采集4 000~400 cm-1范围内的红外谱图。采用北京博渊精准科技发展有限公司生产的热重分析仪进行热重分析,获得SAPO-34分子筛样品的模板剂焙烧温度和分子筛的热稳定性,空气流量为80 mL·min-1,升温速度为10 ℃·min-1,升温范围为室温到700 ℃。采用天津先权公司生产的TP-5079全自动多用吸咐仪进行分子筛酸性(NH3-TPD)分析。采用日本日立公司生产的S-4800型冷场发射扫描电子显微镜观测分子筛的形貌。采用布鲁克生产的Bruker 400M 宽腔固体核磁共振波谱仪上进行27Al-NMR分析,7 mm CP/MAS宽带双共振固体探头,频率约为6 kHz。采用美国康塔Autosrb IQ吸附仪测定比表面积数据,用Brunauer-Emmett-Teller(BET)等温方程计算样品比表面积,采用t-plot方法计算微孔容、总比表面积、微孔比表面积和外孔比表面积。
1.3 MTO催化反应性能评价
采用固定床反应器进行分子筛MTO反应性能的评价,催化反应条件:将焙烧后的分子筛产品压片,筛分出粒径为0.250~0.425 mm的颗粒,装于不锈钢反应器中,以纯甲醇(分析纯)为原料,空速为9.6 h-1,反应温度为450 ℃。气体经收集由德国Bruker公司生产的450-GC型气相色谱仪分析。
2 结果分析
2.1 SAPO-34分子筛XRD表征
图1和图2分别为晶种法和晶种导向液法合成SAPO-34分子筛的XRD谱图和晶化曲线。采用晶种法制备时,晶化4 h,样品在2θ=9.5°、12.8°、15.9°和20.5°就出现了SAPO-34分子筛的衍射峰,但此时衍射峰强度较低,并且有氧化铝的特征峰。随着晶化时间的延长,氧化铝的特征峰不断降低,大约24 h后氧化铝的特征峰完全消失,仅有SAPO-34的衍射峰。继续延长晶化时间,分子筛的结晶度仍会增加,36 h时结晶度基本达到最大值;再进一步延长晶化时间,结晶度不再变化。当溶液中不加晶种时,SAPO-34分子筛一般晶化时间为36~72 h。可见,固体晶种的加入可以明显缩短分子筛的晶化时间[17]。晶种法合成时原料的溶解、晶核的生成、以及晶体的生长是同时发生的。晶种在反应过程中通常是先消溶,然后产生一些相互连接的环状结构,这些结构再进一步重排形成分子筛。晶种或晶种表面脱落的细粉,一方面可以提高晶化体系中的过饱和度;另一方面可以作为晶核,提高晶化速率[18]。
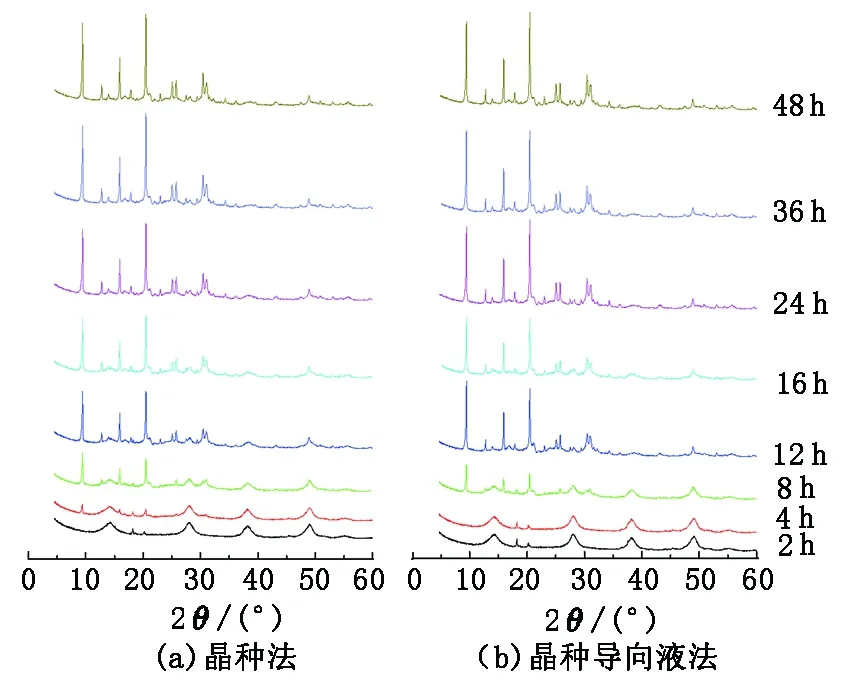
图1 两种方法晶化不同时间制备的SAPO-34分子筛的XRD谱图Fig.1 XRD spectra of SAPO-34 molecular sieves synthesized by two methods for different crystallization time
采用晶种导向液法合成时,整个晶化过程与晶种法的晶化过程基本类似,晶化时间在10 h以下时,两种方法制备样品的结晶度非常接近;晶化时间为12~24 h时,晶种导向液法制备样品的结晶度要高于晶种法制备的样品;晶化时间到36 h时,两种方法制备样品的结晶度又比较接近。两种晶化过程存在如下不同:①晶种导向液法晶化4 h,并没有出现SAPO-34分子筛的衍射峰,仅有氧化铝的特征峰。这说明晶种法直接添加的分子筛晶种,晶种的解体程度不高,因此在部分较大的晶种周围在短时间内就出现了分子筛的晶体。晶种导向液法制备时,凝胶先经过高温晶化1 h,这段晶化时间可以使晶种的溶解更充分、更均匀,并促进了原料的进一步充分反应,形成更多微小的晶体颗粒,因此没有出现短时间内个别晶体生长的现象。②晶种法在36 h分子筛结晶度达到最大值;而采用晶种导向液法,晶化24 h分子筛的结晶度就达到了最大值,继续延长晶化时间,结晶度变化不大。说明采用晶种导向液法尽管在整体上添加的晶种减少,仅为晶种法使用晶种量的1/22,但这少量的晶种制成的导向液中形成了更多的晶核,从而促进凝胶在较短的晶化时间内得到高结晶度的纯相SAPO-34分子筛。

图2 两种方法合成SAPO-34晶化曲线的对比Fig.2 Crystallization curve of SAPO-34 molecular sieves synthesized by two methods with different crystallization time
2.2 SAPO-34分子筛SEM表征
图3为晶种法和晶种导向液法制备的SAPO-34分子筛的SEM。由图3可知,采用晶种法晶化不同时间合成的SAPO-34分子筛的晶粒呈现典型的规整的立方体形貌,晶粒分布均匀、不存在团聚生长的现象。当晶化时间为24 h,平均粒径为1.5 μm,颗粒表面及附近有较多的不规则的小颗粒;晶化时间为36 h,平均粒径为2.0 μm,晶粒粒径略有增大。尽管此时结晶度已经达到最大值,但仍有一些不规则的碎片颗粒,但数量已经明显少于晶化24 h的样品。晶化时间为48 h,样品的平均粒径为1.8 μm,分子筛晶粒的粒径基本变化不大,晶粒表面更加光滑平整,粒度均匀,无晶粒聚集生长现象,晶粒周围的晶体碎片已经非常少。由此认为,采用晶种法合成时,尽管晶化24 h时,就已经形成了分子筛的基本形貌,但有较多的未结合成晶体的碎片,这与结晶度相对较低对应;当晶化时间达到36 h时,尽管结晶度已经达到最大值,但晶体表面不够光滑,仍然存在较多的碎片颗粒;再延长晶化时间至48 h,主要是促进晶体内部结构的完善,以及晶核碎片在颗粒表面缺陷位置的附着生长,不再增加颗粒直径。有文献[19-21]指出,适当的延长晶化时间会促进颗粒的聚集和生长,以及部分小颗粒的溶解和其在大颗粒上的沉积,从而得到较大颗粒的分子筛,所以控制晶化时间可以得到小颗粒的分子筛。但从本文结果可以看出,过短的晶化时间会造成材料中存在较多的碎片,应选择合适的晶化时间。
采用晶种导向液法制备的分子筛的整体颗粒形貌基本与晶种法的相似,都为立方体。但与晶种法相比,采用导向液法制备时颗粒的生长过程明显不同。首先,凝胶晶化16 h制备样品的颗粒表面棱角就已经非常分明,表面已经基本无小颗粒的碎片,表面光滑程度与晶种法晶化48 h的样品非常接近,晶粒粒径约为3.5 μm;继续延长晶化时间至24和36 h,晶粒基本不变,晶粒表面更加光滑,但晶化24和36 h的两个样品的表面光滑程度变化不大。这与晶化曲线基本相吻合,分子筛晶化时间达到24 h,其结晶基本完成,继续延长晶化时间,结晶度基本不变。这说明与晶种法合成相比,采用晶种导向液法晶化,晶体的内部和表面更趋于同时生长,而不是先形成颗粒再进行表面的修饰。所以本实验采用廉价模板剂、简化的晶种导向液法能在更短的晶化时间内合成出高结晶度、表面光滑的SAPO-34。

图3 两种方法晶化不同时间制备的SAPO-34分子筛的SEM图Fig.3 SEM images of SAPO-34 molecular sieves synthesized by two methods for different crystallization time
2.3 SAPO-34分子筛BET表征
表1为不同方法制备的样品的比表面积数据。可以看出,晶种和晶种导向液法制备的分子筛总的比表面积差别不大。晶种法制备的分子筛的外表面积略高:一方面是由于晶种法合成的样品晶粒较小;另一方面从扫描电镜可以看出在晶化的过程中,小颗粒会慢慢生长在颗粒表面,可能在小晶粒重新生长过程中存在一定的颗粒空隙。文献[22]合成的SAPO-34的比表面积一般在435 m2/g,本文中晶种和晶种导向液的加入都可以不同程度地增加分子筛的外表面积,晶种法更有利于增加外表面积;晶种导向液法更有利于增加微孔表面积。

表1 两种方法合成的SAPO-34分子筛的比表面积Table 1 Specific surface area of SAPO-34 zeolites synthesized by two methods
2.4 SAPO-34分子筛DTA表征
在分子筛合成中,模板剂除了具有结构导向和空间填充作用外,还具有平衡分子筛骨架电荷的作用[23],而这些负电荷是由于硅原子进入骨架取代 P原子或 P-Al造成的。因此,模板剂数量也在一定程度上决定了骨架中硅含量。模板剂所能提供的正电荷数决定了分子筛的骨架负电荷数。图4比较了不同方法合成的分子筛的热重曲线,可知两种方法具有类似的曲线,均有3个失重峰,峰型相似。在25~120 ℃之间的峰为吸热峰,是分子筛的物理吸附水脱附造成的;第二个峰是在450 ℃附近强的放热峰,是分子筛孔道中模板剂的氧化分解引起的;大于450 ℃高温区的失重是残留在孔道或笼中的有机物的进一步脱除产生的。两种方法制备的分子筛的模板剂燃烧峰相差不大,说明孔道中模板剂数量基本接近。

图4 两种方法合成SAPO-34分子筛的DTA谱图Fig.4 DTA spectra of SAPO-34 synthesized by two methods
2.5 SAPO-34分子筛IR表征
红外骨架振动可以检测到微量杂晶的存在和骨架的变化,由图5的红外谱图可知,不同方法合成的分子筛的振动峰峰型相似,与SAPO-34分子筛的特征骨架振动峰吻合[24-25],并没有发现SAPO-34分子筛以外的骨架振动峰,说明合成方法的改变并不影响分子筛的纯度。480 cm-1处的SiO4四面体振动峰和640 cm-1处的双六元环特征振动峰,表明CHA结构SAPO-34分子筛的生成。530 cm-1处为AlO4或SiO4四面体的T—O弯曲振动峰,575 cm-1处为PO4四面体的T—O弯曲振动峰;730 cm-1处为O—P—O或O—Al—O对称振动峰;1 100 cm-1处为O—P—O非对称振动峰;1 215 cm-1处为P—O—Al或P—O—P非对称振动峰。两种方法合成的样品在480 cm-1处振动峰略有偏移,说明硅的分布略有差别。

图5 两种方法合成SAPO-34分子筛的红外谱图Fig.5 FT-IR spectrum of SAPO-34 zeolites synthesized by two methods
2.6 SAPO-34分子筛NH3-TPD表征
由表2可知,不同方法合成的分子筛都具有强酸和弱酸两种酸类型,但酸量略有差别。与晶种法比,导向液法合成样品的总酸量略小,主要是弱酸量较少。二者的强酸性的峰温位置变化不大,峰面积差别不明显。可见在相同的原料和投料硅铝比的条件下,改变合成方法调整晶体的调节生长过程,可以调变分子筛的酸性分布。从晶化过程来看,晶种法更趋向于先形成晶体再进行后期的晶面和晶体内部的完善,在晶面完善的过程中是硅取代的主要阶段;而晶种导向液法合成时,晶体的完整性几乎在短时间内完成,这个过程中硅磷铝键几乎同时生成,更易形成独立的硅原子[26],降低酸性。

图6 两种方法合成的SAPO-34分子筛的NH3-TPD图Fig.6 NH3-TPD results of SAPO-34 zeolites synthesized by two methods

合成方法弱酸酸量/(mmol·g-1)强酸酸量/(mmol·g-1)总酸酸量/(mmol·g-1) 晶种法0.320.160.48 晶种导向液法0.260.170.43
2.7 SAPO-34分子筛27AL核磁共振表征
B酸位的产生来自于Al和Si之间形成的桥羟基,因此铝的环境也与酸性质有关。在化学位移δ=40×10-6和δ=65×10-6附近的归属于具有Al(OP)4结构的骨架4面体的铝。δ=-10×10-6附近处峰归属为6配位的铝[27-30]。5配位和6配位的铝来源于铝与骨架外的水、羟基相互作用或非骨架铝。说明两种方法合成的分子筛铝配位环境相差不大,这与
酸性差别不大。δ=10×10-6附近的峰归属为未反应的铝源或4配位的铝与水或模板剂形成的5配位铝。
2.8 甲醇转化性能
将不同方法制备的样品进行了甲醇转化反应性能的对比,结果见表3。在相同的反应条件下,导向液法合成的样品具有略低的转化率,这可能是因为该样品具有相对较弱的酸性和略大的晶粒。然而,晶种导向液法制备的样品具有较高的烯烃收率,与晶种法相比,乙烯和丙烯的收率总和高出3%,选择性高出4%。这与文献中所提到的较弱的酸性更有利于烯烃的生成规律是一致的[31-32]。说明导向液法合成的样品更有利于提高烯烃的选择性。

图7 两种方法合成的SAPO-34分子筛的Al核磁谱Fig.7 Al-NMR results of SAPO-34 zeolites synthesized by two methods
3 结束语
以廉价的三乙胺为模板剂、以最廉价的硅溶胶和拟薄水铝石为硅源和铝源、采用简化的晶种导向液法制备低硅铝比SAPO-34分子筛(硅铝比为0.2),不仅大大简化了现有的晶种导向液法合成程序和成本,晶种的用量也大大降低,仅为晶种法合成时用量的1/22,而且晶种导向液法改变了分子筛的晶化过程,具有更短的晶化时间,晶化时间仅为24 h,至少缩短了8 h;晶粒略大,约为3.5 μm,增加1.5 μm;微孔表面积较大;酸性略低和转化率略低,但烯烃收率高出3%,选择性高于4%,更适合用于MTO转化过程。
猜你喜欢
黑龙江大学自然科学学报(2022年1期)2022-03-29
煤气与热力(2021年9期)2021-11-06
陶瓷学报(2021年1期)2021-04-13
发明与创新(2020年31期)2020-12-20
军事文摘(2020年20期)2020-11-16
石油炼制与化工(2020年8期)2020-08-06
丝绸(2020年6期)2020-06-23
中学生数理化·八年级物理人教版(2020年12期)2020-01-01
江苏农业科学(2015年1期)2015-04-17
科技创新导报(2014年34期)2015-01-13
