
全基因组关联分析的研究现状及对数据科学的挑战
2019-05-07 01:46
广州大学学报(自然科学版) 2019年1期
(西安电子科技大学 计算机科学与技术学院, 陕西 西安 710071)
全基因组关联分析的文章很多,生命体的复杂性,使得全基因组关联分析涉及的问题方方面面,本文则重点从数据挖掘和数据科学的角度,探讨全基因组关联分析的现状、问题及其对数据科学的挑战.
1 关于GWAS
1.1 GWAS的定义
全基因组关联分析(Genome wide association study, GWAS)是通过考察全基因组范围DNA变异的单核苷酸多态性(SNP),挖掘影响复杂疾病等的表型性状(如疾病、癌症、身高等)的SNP的计算方法.
孟德尔疾病,又称单基因病,是由一对等位基因控制的疾病或病理性状,人体中只要单个基因发生突变就足以致病的一类遗传性疾病.而非孟德尔疾病,亦称复杂疾病(complex disease),为多基因病,即疾病或病理性状是多个基因、它们的交互作用、以及它们与各种环境因素的交互作用所导致的联合效应的结果.人类目前面临的未解疾病大多属于复杂疾病,包括各类神经性疾病(癫痫、癔病、神经分裂症、阿尔茨海默病等)、各类肿瘤和癌症、糖尿病、心脏病、骨质疏松症、哮喘以及各类疑难杂症等,以及各类表型性状(如骨密度、支气管反应性、葡萄糖水平、对环境刺激的反应如药物功效或副作用)等,如何认识其致病机理是21世纪生物学和生物医学的重大挑战.
所谓SNP主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性,它是人类可遗传的变异中最常见的一种,占所有已知多态性的90%以上.SNP在人类基因组中广泛存在,平均每500~1 000个碱基对中就有1个,其总数可达百万到千万数量级[1].
2005年Klein等[2]发表在Science上的文章,第一次成功鉴定了影响年龄相关性黄斑变性病的重要遗传因子,这之后GWAS研究的范围越来越广泛.
1.2 GWAS所面临的数据:超高通量超小样本超大噪声

由于全基因组中涉及的SNP达到百万甚至千万数量级,也就是说xi的维度达百万甚至千万,甚至生物学家也无法告知导致疾病的DNA变异可能是哪些,这正是生物学家和生物医学家向数据科学家提出的问题:数据科学家能否通过对上述数据的处理,从数据中挖掘出与疾病关联的SNP位点,这是生物学家向数据科学家提出的严重挑战.之所以要从全基因组范围中挖掘,是为了不漏掉任何可能的致病SNP,因为生命和疾病现象极其复杂,任何SNP都可能是疾病的根源.
其实,DNA作为遗传密码,其单核苷酸多态性(SNP)正好反映了人类个体间的性状差异和个体的多样性,诸如身高、体重、胖瘦、体态、各类疾病等,随着研究的深入,GWAS的数据越来越大(样本越来越多),将导致除了上述个体多样性外,还存在由于地域、人种、群体等不同所带来的样本的群体多样性.标识出有病和没病或表型性状,只是为了从如此众多的SNP中找出仅与疾病/表型性状关联的SNP,而将所有其他的多样性均视为是对其的干扰和噪声.
1.3 GWAS的研究目标
全基因组关联分析对数据分析的目标,是要从全基因组的所有SNP中,找出仅仅与疾病相关联的SNP,从科学上讲,它是为科学服务的——为生物学家提供与疾病相关的科学发现和研究疾病发生发展机理服务;从临床应用上,针对这些SNP进行疾病/癌症的早期诊断;从研发上,针对所发现的与疾病关联的SNP,将其作为药物靶点进行分子药物研究和对疾病/癌症的早期治疗研究.
实际上,GWAS更加关注多SNP联合致病,亦称SNP的交互作用,而对于某个特定疾病,到底有多少个SNP联合致病、是哪几个SNP的联合致病、以及这几个SNP联合起来是怎样致病的,这些都是GWAS需要从数据中挖掘和解决的问题.
本文认为,GWAS的最终目标,应该是从全基因组的所有SNP中,找出复杂疾病的分子致病原因,而不是找出与复杂疾病仅仅是相关联的SNP,因为只有找出真正的致病原因,才能引发生物学家的研究发现,而错误的发现将误导他们的研究发现,误导对复杂疾病机理的认识.只是目前的技术手段有限:由于对“原因”这个问题还无定性和定量的科学表述,不得已找与复杂疾病相关联的SNP罢了.
2 研究现状——数据、方法及成绩
GWAS研究,无论在数据、方法和对肿瘤/癌症的研究上,都已取得了可喜的成绩.数据上,各国出台了相应的大规模甚至超大规模的基因组计划,为疾病相关GWAS研究奠定了数据基础;方法上,更多采用统计学方法、机器学习和智能优化等方法.
2.1 数据上的进展
WTCCC(Wellcome Trust Case Control Consortium,https://www.wtccc.org.uk/)从2005年开始就建立了全基因组SNP样本数据库,为研究者提供了很好的数据平台[2].癌症基因组(TCGA)计划(http://cancergenome.nih.gov/)整合了不同芯片平台的多层面数据,为识别癌症相关的生物标记提供可能.目前,TCGA中包含了30多种癌症和肿瘤的数据,每种疾病包含了基因组(DNA序列层面的变异)、表观遗传组(DNA甲基化和miRNA表达)和转录组(基因表达),为研究疾病的内在致病机理提供可能.国际千人基因组计划(http://www.1000genomes.org/),通过解码1 000多来自非洲、亚洲、欧洲和美洲共14个民族的基因组,绘制出迄今为止最大最全的人类遗传变异目录.该计划最终将解码26个民族群体共2 500个人的基因组,寻找世界各地人群中复杂疾病的遗传基础.此外还有“炎黄计划”(包含南北方地区的中国人)、“中国新生儿基因组计划”和“中国胚胎基因组计划”等.2017年12月28日,我国启动“中国十万人基因组计划”,这是我国在人类基因组研究领域实施的首个重大国家计划,覆盖地域包含我国主要地区,涉及人群除汉族外,还包括人口数量在500万以上的壮族、回族等9个少数民族.这些都为通过GWAS解密复杂疾病的遗传密码,奠定了良好的数据基础.此外,英国10万人基因组计划、美国的100万人个人健康信息以及基因组测序、韩国万人基因组计划、法国基因组和个体化医疗计划(法国基因组医疗2025)等也正在各国展开.这些计划的实施,为研究人类健康和各类遗传疾病的遗传机理、发现疾病的分子致病原因和生物学家的生物学发现、分子靶向药物研制和疾病早期诊断和个性化治疗,提供了良好的数据基础.
GWAS就是要回答在百万到千万数量级的SNP中哪些SNP是与疾病真正关联的问题,那么如果用某种方法找到了答案,该答案是否正确,需要进行昂贵的生物实验来验证,为此需要在此之前先进行计算评价.目前有2种形式进行计算评价:①通过对数据的仿真,在仿真数据中嵌入相应的答案,考察数据挖掘的结果是否正好是所嵌入的答案;②在没有答案情况下(真实数据即为这种情况),考察所获得解的统计重要性.
2.2 研究方法的进展
在研究方法上,统计方法、信息论方法、机器学习和深度学习方法、智能优化方法等均已运用到GWAS研究中.
早期的研究针对的数据集较小,更多采取穷举法,比如组合划分法(combinatorial partitioning method, CPM)[3]、多因子降维(multifactor dimensionality reduction, MDR)[4]、受限组合划分法(restricted partitioning method, RPM)[5]、信息增益法(information gain)[6]、反向基因型-表型关联法(backward genotype-trait association,BGTA)[7]等,这些方法通常不适于大规模数据集的GWAS.
鉴于全基因组涉及的SNP数目巨大,找仅与表型相关的SNP更多的是分2步甚至多步进行,首先滤除那些认为与表型只有微不足道关联的SNP,再在剩余的SNP中搜索与表型关联的SNP.通常滤除过程采用轻量级计算,而在剩余SNP中搜索则更多采用诸如穷举的重量级搜索和计算,以保证搜索过程能在有限时间内高效完成.目前已开发了许多随机和启发式方法,这些方法有可能保留尽可能多的具有表型信息含量的SNP,同时极大地降低计算的复杂度从而适应大规模数据集.
例如,Tang等[8]提出的epistatic module detection (epiMODE)是基于蒙特卡洛随机抽样策略的贝叶斯推理算法BEAM的推广;Wang等[9]提出了AntEpiSeeker,它是一个2步的蚁群优化搜索算法;Wan等[10]提出了基于预测规则推理和2步设计的SNPRuler,他们还提出了另一种方法,基于布尔操作的筛选测试方法BOOST[11],其中仅涉及布尔值,并允许使用快速逻辑操作以获得列联表.文献[12-14]提出了一系列方法,利用所采用的测试统计量的性质来缓解多个测试问题,其中基于树的关联分析方法(TEAM)通过使用最小生成树来更新两位点的列联表.
近年来,深度学习也应用于GWAS.Paul等2018年提出了识别高阶SNP交互作用的新方法,该方法基于SNP组合的非线性变换,利用原始SNP序列的变换域表示对深度学习分类器进行初始化,利用自编码器来识别高阶SNP交互作用[15].
为检验与肥胖表型相关的统计显著的单核苷酸多态性(SNP)的预测能力,Montaez等[16]展示了深度学习作为GWAS分析的潜力,该框架可以捕获关于SNP的信息以及它们之间的重要交互作用,结果表明,运用深度学习可以捕捉到单一的SNP分析不能捕捉到的不太显著变异的累积效应,及其对疾病预测结果的总体贡献.
然而,Bellot等[17]在Genetics上的论文则指出,深度学习(DL)在复杂人类性状基因组预测中的性能还没有得到全面的测试.在其所评估的性状范围内(身高、骨密度、体重指数、收缩压和腰臀比5种表型),卷积神经网络(CNN)的表现与线性模型的基本相当:没有发现DL以相当大的幅度超过线性模型的情况[17].
理想的GWAS数据,其群体中的个体彼此的差异度应该是相同的,个体间唯一最大的差异应该是与控制目标性状的基因之间的差异.但实际情况往往并非如此,这就是所谓的群体多样性.为去除GWAS数据中群体多样性的影响,目前的主要方法是基因组控制法(通过修订关联统计量)[18]、分层分析法(利用非关联的分子标记和贝叶斯聚类技术)[19]、主分量分析法(计算亲缘关系矩阵的特征值和特征向量)[20]、混合线性模型法(将表型分解成固定效应、随机效应和残差效应的线性叠加)等[21],以降低由于样本的群体结构所导致的GWAS偏差.
2.3 取得的成绩
GWAS研究已取得了很好的成绩.例如,Klein等[2]利用GWAS进行老年黄斑变性疾病(Age-Related Macular Degeneration, AMD)的研究,从116 204个SNP位点中,发现了2个位于基因CFH内含子上的SNP位点(rs380390,rs1329428)与AMD具有很强关联性.Cai等[22]从22 780个case样本和24 181个Control样本中发现了30个独立的乳腺癌易感基因位点.文献[23]通过GWAS研究发现TERT-CLPTM1L位点的常见变体与雌激素受体阴性乳腺癌相关.2017年,Michailidou等[24]对122 977名欧洲乳腺癌患者和105 974名对照者进行了基因分型阵列和SNP分析,在已知的和新的风险位点之间发现了潜在联系,证实了许多之前发现的乳腺癌致病位点,并找出了65个新的乳腺癌致病基因位点,证明了乳腺癌候选靶基因和体细胞驱动基因之间有很强的重叠.Milne等[25]研究了21 468个雌激素受体阴性乳腺癌患者,通过GWAS分析与雌激素受体阴性乳腺癌风险的关联性,证实了之前发现的10个易感基因位点.2018年, Lillias等发现了39个SNP位点与憩室病(diverticular disease)紧密相关[26].
身体质量指数(BMI)和血清胆固醇等健康风险因素与许多常见病有关.Zhu等[27]开发并应用了一种方法(称为GSMR),用全基因组关联研究的汇总数据进行多SNP孟德尔随机化分析,研究了BMI、腰臀比、血清胆固醇、血压、身高和受教育年限等与常见疾病(样本数量达405 072个)之间的因果关系,包括低密度脂蛋白胆固醇对2型糖尿病(T2D)的保护作用和二型糖尿病他汀类药物的副作用,以及EduYears对阿尔茨海默症的保护作用等.
2018年10月,英国牛津大学的Stephen带领的研究组在Nature Genetics发表文章,他们利用UK Biobank数据库提供的共8 428个样本的全基因组测序以及多模态脑影像数据,对共3 144个功能和结构脑影像指标进行了全基因组关联分析,发现了148个可重复的由SNP和其相关脑影像指标组成的簇, 发现与铁转运和储存相关的基因与皮质下脑组织磁化率有关,还有17个相关基因同大脑的发育、信号通路以及可塑性相关[28].
Huyghe等[29]2019年发表在Nature genetics的论文,对1 439例病例和720例对照者进行了全基因组测序,将发现的序列变异和单倍型纳入全基因组关联研究数据,并对34 869例病例和29 051例对照进行了相关性测试.另有23 262例病例和38 296名对照者对调查结果进行了跟踪.遗传力分析表明,结直肠癌风险是高度多基因的相关的,更大、更全面的研究能够进行罕见的变异分析,并将提高对这种风险背后的生物学的理解和影响个性化筛选策略和药物开发.
Chimusa等[30]开发的ancGWAS是一种基于代数图中心性的方法,通过将GWAS数据集的关联信号集成到人类蛋白质-蛋白质交互作用(PPI)网络中,考察在识别重要疾病子网络中的连锁不平衡,从而尽可能地消除复杂疾病发病机制背后的基因之间的相互作用,为疾病的发病机理提供进一步的认识.
3 面临的挑战——问题极其严重
Ioannidis[31]早在2005年发表在PLoS Medicine上的论文标题——“为什么大部分的研究发现是错的”,已明确指出了存在问题的严重性.Park等[32]也指出:在不值得注意的GWAS结果中有许多是值得关注的,这是由于样本集大小的影响.本文则认为,样本大小只是问题的一个方面,其背后有着更深层次的原因.
3.1 GWAS结果的重复性差
以一个典型数据的GWAS研究结果来看这个问题.以年龄相关性黄斑变性(Age-related Macular Degeneration,AMD)为例,这是GWAS最早研究的疾病,研究最早、最广泛、最充分,这里仅列出3个代表性的研究结果,可以看到这些结果的不一致性(即重复性差),并且结果中的重叠性高.
Jiang等[33]2009年发表在BMC bioinformatics上的论文,在对SNP进行初步筛选的基础上,运用具有100万个决策树的超大规模随机森林,通过暴力搜索(学习),获得表1的结果(其中的P值未经Bonferroni校正).
表1文献[33]给出的对AMD数据进行GWAS的结果
Table 1 Results of GWAS on AMD data presented in literature [33]

SNP InteractionP-value(rs6104678,rs7863587)1.28×10-7(rs3743175,rs1394608)3.06×10-7(rs2828155,rs1394608)3.06×10-7(rs4292478,rs1394608)7.29×10-7(rs6104678,rs10512174)7.68×10-7(rs2347060,rs3758141,rs7104698)5.57×10-9(rs2347061,rs3758141,rs7104698)5.57×10-9(rs2347060,rs10503640,rs7104698)6.91×10-9(rs2347061,rs10503640,rs7104698)6.91×10-9(rs2347060,rs1557753,rs7104698)1.07×10-8
2017年发表在Scientific Report上的论文[34],搜索数据中存在的3个SNP联合致病的因子,得到表2的结果(其中的P值也未经Bonferroni校正).
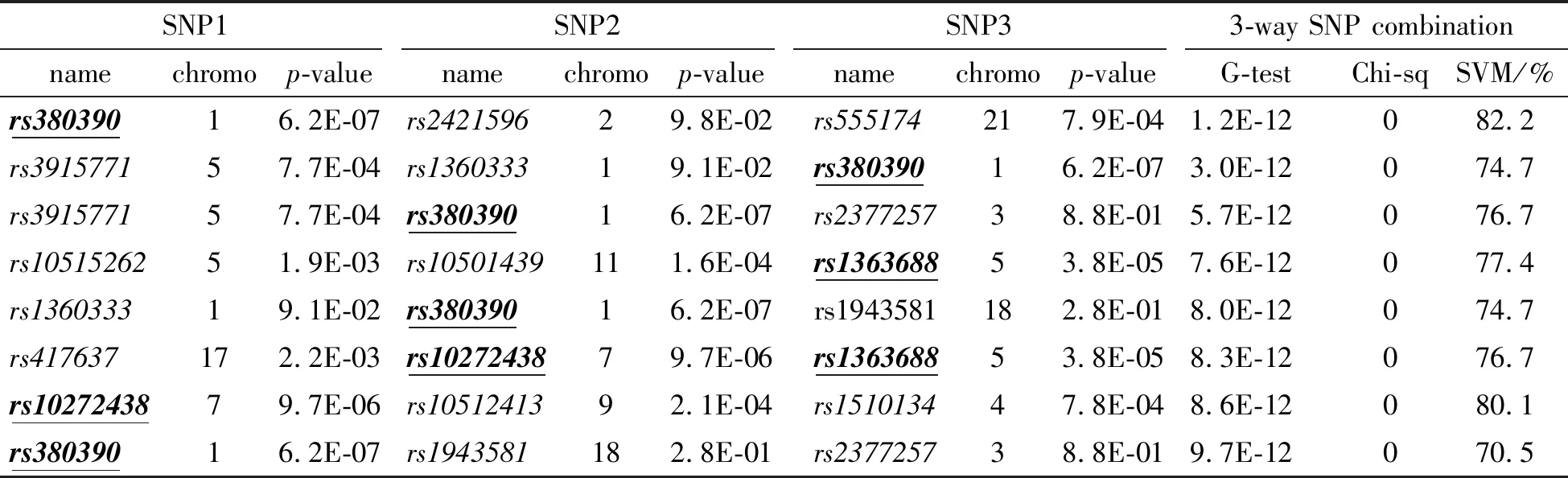
表2 文献[34]给出的对AMD数据进行GWAS的结果
2016年发表在BMC Bioinformatics上的论文[35],则用图示方式更鲜明地表达了类似的重叠现象,见图1(其中黑色表示单个SNP的边缘效应,灰色表示2个SNP的联合效应,淡灰色表示3个SNP的联合效应;圆圈的面积越大,表明效应越强).

图1 文献[35]给出的对AMD数据进行GWAS的结果
Fig.1 Results of GWAS on AMD data presented in literature [35]
从上述结果看出:①相同数据,但结果都不一致,亦即结果没有重复性;②找到的SNP组合,存在严重的重叠结构,如表1中的rs6104678在2个SNP的组合(rs6104678, rs7863587)和(rs6104678, rs10512174)中存在,类似重叠现象在上述表1、表2和图1中均可见到.显然,重叠表明所找到的不是本质原因,如果重叠,则其背后必有更本质的原因,而GWAS的目标是找出复杂疾病/表型性状的本质原因;③超大规模位点数量,使得无法对结果运用多重检验对P值进行修正;④诸如随机森林、ROC曲线等,均为分类器或评价分类器的评价指标,却未必能够评价本质原因.
3.2 GWAS对数据科学的挑战
通过GWAS找表型的本质分子原因,是生物学和生物医学向数据科学提出的严峻挑战.
3.2.1 从数据中到底要挖掘出什么并不明确
关于全基因组关联分析,其目标是从数据中挖掘出与疾病关联的SNP,然而,关联在这里到底应该是怎样的含义并没有严格给出.关联可以认为是一种联系,但是从一般意义上讲,任何位点都有联系,那么怎样的联系算是关联?这里是要找联系,还是本质原因?本质的含义到底是什么?原因的含义又是什么?这些问题实际上尚未厘清.结果是,目前从数据中挖掘出的,更多的是只具有统计重要性的SNP,而统计重要性未必是科学重要性.
似乎,GWAS的目标可以认为是从数据中挖掘出“真正”致病的SNP“原因”,那么什么是“原因”,什么是“真正的”,这些看似有点哲学味道的问题,实际上已经困扰哲学界上千年,显然在计算上目前尚未解决.
3.2.2 研究发现不容忍差错
GWAS是通过数据分析的手段,从数据中挖掘出疾病的致病SNP的,以便生物学家进一步地认识疾病机理,因此,任何错误的发现,都会误导生物学家对疾病机理的认识,误导生物学的研究发现.这个问题与人们生活中大量存在的模式识别问题(比如人脸识别问题,指纹识别问题等)是完全不同的,在这些模式识别问题中,只要模式被正确识别就算达到目的,并不探究更深层次的机理问题.比如人脸识别问题,只要能正确识别就行,至于到底是人脸的哪些差异能最好地帮助识别,并不关心.因此,所有模式识别方法甚至机器学习方法,对全基因组关联分析可能是无益的,所发现的可能离真正的致病原因相差甚远,尤其在小样本情况下可能更是如此.
3.2.3 将GWAS问题当做统计学和机器学习问题
比如,Xu等[36]将GWAS视为一个特征选择问题,Wei等[37]对约13 000 个溃疡性结肠炎和约22 000个对照组的样本,通过运用各种机器学习的技术进行GWAS,获得了0.83的AUC性能,而AUC通常是衡量分类算法性能的指标.将GWAS问题当做统计学和机器学习问题的研究比比皆是,常常将疾病的分子预测与疾病的分子致病原因混为一谈,然而从GWAS数据中挖掘表型性状的分子原因,与挖掘统计重要性(统计学方法)、挖掘出模式(机器学习方法)、挖掘出对疾病的预测风险等,应该是截然不同的目标和方向.
目前的机器学习,都是在假设空间中学习[38],比如深度神经网络,其网络的结构本身就已设定了假设空间,而众多参数的超高维空间学习问题,则是在这个超高维空间中的超大规模搜索问题,对于这个搜索问题,目前的智能优化技术(比如遗传算法、蚁群算法、和声算法等),仍难于保证搜索到全局最优解.然而,学习的目标应该是保证什么最优呢?回答是保证推广能力最优(强),即用对训练数据(seen data)处理所获得的知识(尽管难于解释)来最大限度地处理新的没有见过的数据(unseen data).由于目前还未能对所谓的推广能力有更加科学的表述,在目标函数上目前更多的是针对训练数据的实际输出和期望输出的误差平方和最小来设定,或以用交叉验证的平均识别率最大来设定.然而,这些目标函数实际上并不能真正表征学习目标是否达成,因为与误差平方和最小相伴的有可能是过拟合,与交叉验证的平均识别率最大相伴的有可能是交叉验证各识别率的波动,从而平均识别率实际上可能只是一个偶然现象,2者实际上都是不能真正检验对未见过的数据的处理性能的.实际上,即使用统计上的P值评价,也同样存在P值的波动现象,尤其在小样本情况下更是如此.
因此,不应将疾病的分子预测与疾病的分子致病原因混为一谈(尽管它们可能会是有联系的),目前的机器学习技术似乎还不足以从GWAS数据中挖掘出表型性状的分子原因;即使可以,建立在统计机器学习基础上的GWAS也存在着诸如假设是否合理、目标函数设定是否合适、性能评价是否到位等诸多问题,以及全基因组分2步搜索时评价指标(第一步为轻量级计算,第二步为重量级计算)的不一致性、不同方法对致病模型搜索能力存在偏好等[34],阻碍了对真正致病SNP的挖掘.
3.2.4 统计重要性不等于科学重要性
统计学意义的交互作用与生物学意义的交互作用的争论由来已久,其分歧在于:统计学意义的交互仅能给出“可能”是交互作用的结果,而不能回答到底“是不是”真正存在的交互作用[39].本文认为,真正的SNP的交互作用——真正存在的分子致病原因,才是具有科学重要性的,而统计重要的未必就科学重要.即使将统计学意义的交互作用阐述得再好,也无法回答生物学意义的交互作用.从统计学意义的交互作用本身来论证生物学意义的交互作用,是不合逻辑的.
但是目前的绝大多数生物信息学研究(也包括GWAS)的计算评价都是统计重要性,以P值评价及其confidence interval来评价,而由于“真正”的致病“原因”中所谓的“真正”和“原因”还没有获得深入的认识、科学的定义和计算上的解决,目前的研究只能追求统计重要性,而非科学重要性.其结果正如2005年Ioannidis[31]在国际知名刊物PLoS Medicine上发表的、至今已被引用6 600多次的论文,“为什么大部分的研究发现是错的”(Why most research findings are false).究其原因,是所获得的结果不可重复.然而,可重复的发现才是真正科学的发现:重复性是科学的灵魂(2014年发表于《细胞》子刊Chemistry & Biology上的题为“Credibility and reproducibility”的社论的第一句话)[40].2015年发表在Genes and Diseases上的新闻视角,题为“Authentication of experimental materials: A remedy for the reproducibility crisis?”的文章[41],将目前研究发现的不可重复问题称为“重复性危机”.因为“重复性危机”,甚至有人2008年发表论文,怀疑重复性是否是考察基因关联发现的金标准(题目为:Is replication the gold standard for validating genome-wide association findings)[42].然而本文认为,重复性显然是研究发现的金标准,只是“到底要通过研究发现什么(而不是怎样发现)”这个问题,目前还没有技术上的解决方案.
3.2.5 多基因联合致病却可能没有主效应基因
多位点联合对表型起重要作用的认识由来已久,即确定复杂疾病生物特征的是基因的组合.尽管人们已经发现在DNA与复杂疾病如心血管疾病、糖尿病、乳腺癌、肥胖、哮喘、常见的神经系统疾病如帕金森病、癫痫等疾病之间存在着重要的关联关系:①它们与单基因遗传的致病因素不同,虽每个基因对表型的作用是微小的,甚至根本不存在,但他们的组合却可以致病,即它们往往没有足以致病的主基因,因此很难用连锁分析的方法来克隆其致病基因;②如果把这种某位点集合的联合效应视为复杂疾病易感的一个因素(原因)的话,那么人们已越来越认识到复杂疾病易感的原因可能有多个,认识复杂疾病的所有原因,无论对认识疾病产生的机理,还是对疾病的预测、诊断和药物研究,都至关重要.
在全基因组范围对复杂疾病/表型的致病原因的搜索,是一个典型的组合爆炸问题.首先,到底有多少个SNP(记为k)与疾病表型关联是未知的和要从数据中挖掘的,即使这个数目k是已知的(比如k=3),那么,从百万到千万数量级的SNP中(如100万)搜索出这k个SNP,则是在100万的k次方的空间中搜索.也正是如此,对于如此超大规模的搜索空间中搜索到的解的评价,大部分文献中报道出来联合位点的P值甚至无法进行多重检验,从而即使P值很小也未必统计重要,更没法谈科学重要性了.
4 总 结
全基因组关联分析不断发展,数据已从单基因组学数据发展到多组学数据,从单一表型发展到多个表型,从常规变异发展到罕见变异,从复杂疾病的致病分析发展到复杂疾病的发生发展过程分析,从复杂疾病的分子关联分析发展到药物的分子效应分析,从人类性状的GWAS发展到动植物的GWAS等,并均取得了可喜成绩:发现了一些与表型性状具有较强关联的变异,但对复杂疾病的可解释性依然很低,并且结果的可重复性难于保障.不可避免地,样本数量有限是原因之一,但非主要原因.本文认为其主要原因是:①对要解决的问题,目前的表述并不明确(什么叫关联);②解决问题的途径更多的是模式识别、机器学习方法(模式分类、回归分析、聚类分析信息熵等)和智能优化方法;③评价更多的是统计重要性评价或疾病风险预测能力评价(分类性能).上述这些可能离GWAS的研究目标——从GWAS数据中找出“真正”致病的分子“原因”,还有较大距离.然而本文认为,这些问题的解决才是使数据挖掘技术真正成为数据科学的关键.
猜你喜欢
新世纪智能(数学备考)(2021年9期)2021-11-24
今日农业(2021年11期)2021-08-13
中西医结合肝病杂志(2020年2期)2020-10-27
当代陕西(2019年15期)2019-09-02
中成药(2018年7期)2018-08-04
学苑创造·A版(2018年11期)2018-02-01
现代园艺(2017年21期)2018-01-03
读者(2017年5期)2017-02-15
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
