2种PRODAN衍生物分子激发态氢键的理论计算
2019-06-19 07:39赵丹,刘洋,赵震,3
沈阳师范大学学报(自然科学版) 2019年2期
赵 丹, 刘 洋, 赵 震,3
(1. 沈阳师范大学 化学化工学院, 沈阳 110034; 2. 沈阳师范大学 能源与环境催化研究所, 沈阳 110034;3. 中国石油大学 重质油国家重点实验室, 北京 102249)
氢键是一种特殊的弱静电相互作用,大量研究表明氢键作用可以影响分子系统的光化学行为[1-5]。赵广久等[3]研究发现分子间氢键作用在激发态增强,而激发能降低,则氢键复合物的吸收光谱相比其单体分子将发生红移,相反则将发生蓝移。因此,氢键对探究光化学过程具有重要的意义。1979年,Gregorio Weber最先合成了PRODAN(6-丙酰基-2-二甲基氨基萘)[6],它是一种微摩尔探针,对有机溶剂有较强的荧光响应并且对氢键作用有很高的灵敏度[7-12]。
在许多实验中,人们使用荧光探针来检测物质[13]。2016年,Alty等[14]在实验中合成了6种新型PRODAN衍生物分子,为了探究氢键对PRODAN衍生物分子的作用机理,在Alty的实验工作基础上,选取了PRODAN衍生物分子的1a和3a分子作为研究对象,分子结构如图1所示。
为探究1a和3a分子分别在气相和甲醇溶液中的激发态氢键,首先优化了PRODAN衍生物1a和3a分子单体结构,再继续优化其单体在甲醇溶液中所形成氢键复合物的结构,然后通过对几何结构、电子光谱和氢键结合能的对比分析,从而对分子间氢键对激发态性质的影响进行讨论。
1 计算方法
在Gaussian 09程序包[15]下,所有计算均选择了CAM-B3LYP泛函[16]和6-31G**基组,并分别采用DFT和TDDFT方法对分子体系基态和激发态的几何结构进行了优化。同时,在相同水平上计算了氢键结合能、振子强度及电子光谱。另外,选用SMD溶剂模型来计算分子在甲醇溶液中的溶剂化效应。
2 结果与讨论
2.1 基态和第一激发态几何构型优化
为了确定分子间氢键作用对1a和3a基态构型的影响,首先优化了1a和3a分子单体以及其在甲醇溶液中所形成的氢键复合物在基态(S0)的空间构型,如图2所示。
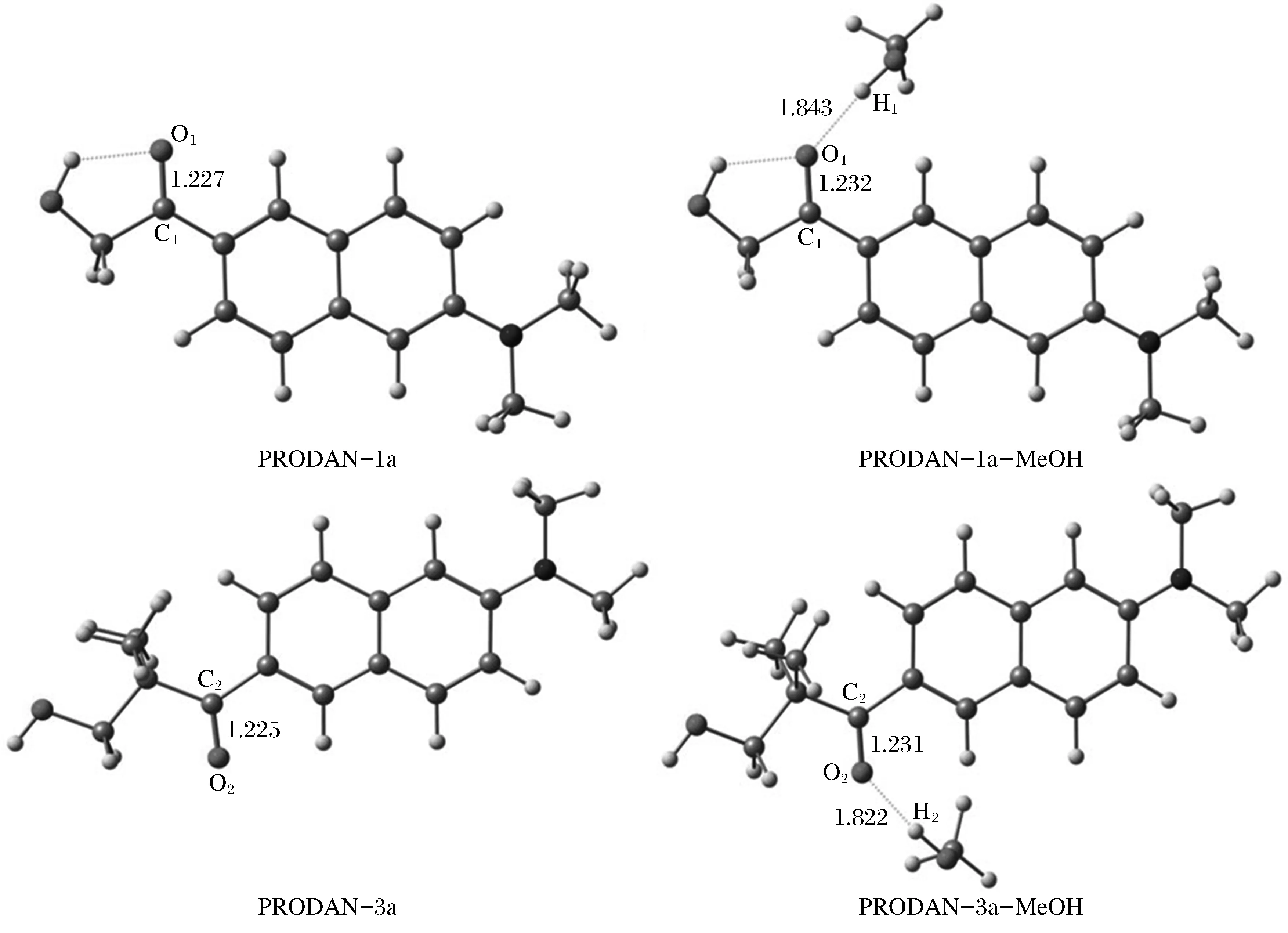
图2 优化后的PRODAN衍生物(1a和3a)及其氢键复合物在基态的分子结构
在基态的稳定构型基础上,优化了1a和3a分子单体以及其氢键复合物在第一单重激发态(Sl)的几何结构,如图3所示。
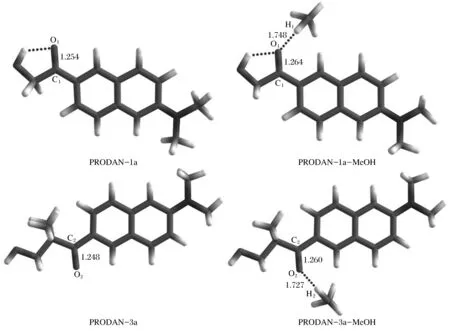
图3 优化后的PRODAN衍生物(1a和3a)及其氢键复合物在激发态的分子结构
计算所得的基态和激发态的键长列于表1。对于气相中的PRODAN衍生物1a和3a分子单体,羰基的键长分别为1.227和1.225 Å;光激发后,对应的羰基键被拉长,分别变为1.254和1.248 Å。在甲醇溶液中,氢键复合物PRODAN-1a-MeOH分子中羰基和氢键O1-H1的键长分别为1.232和1.843 Å;当分子发生激发后,羰基的键长伸长到1.264 Å,而氢键O1-H1的键长则缩减为1.748 Å。对于氢键复合物PRODAN-3a-MeOH分子,羰基和氢键O2-H2的键长分别为1.231和1.822 Å;光激发后,羰基的键长增大到1.260 Å,氢键O2-H2的键长缩减到1.727 Å,通过对键长的变化分析,甲醇溶液中的PRODAN衍生物分子激发态羰基的键长明显拉伸,氢键的键长明显缩短,说明在甲醇溶液中激发态氢键复合物的分子间氢键作用显著增强。
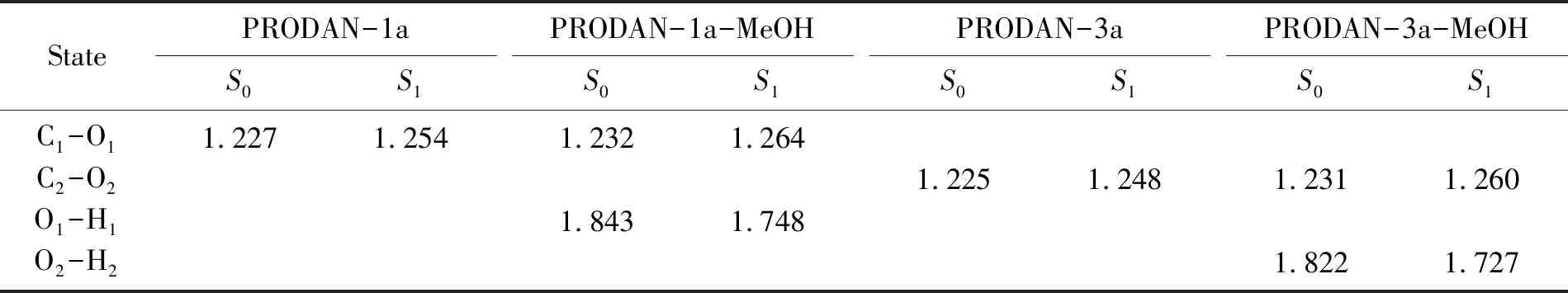
表1 计算得到的PRODAN衍生物(1a和3a)及其氢键复合物的主要键长( Å)
2.2 电子光谱
为了进一步研究激发态氢键作用对PRODAN衍生物1a和3a分子在气相和甲醇溶液中的影响,计算了PRODAN衍生物1a和3a分子单体及其氢键复合物的6个低能电子激发态,并得到相应的吸收光谱和发射光谱,所得的相关数据列于表2中。
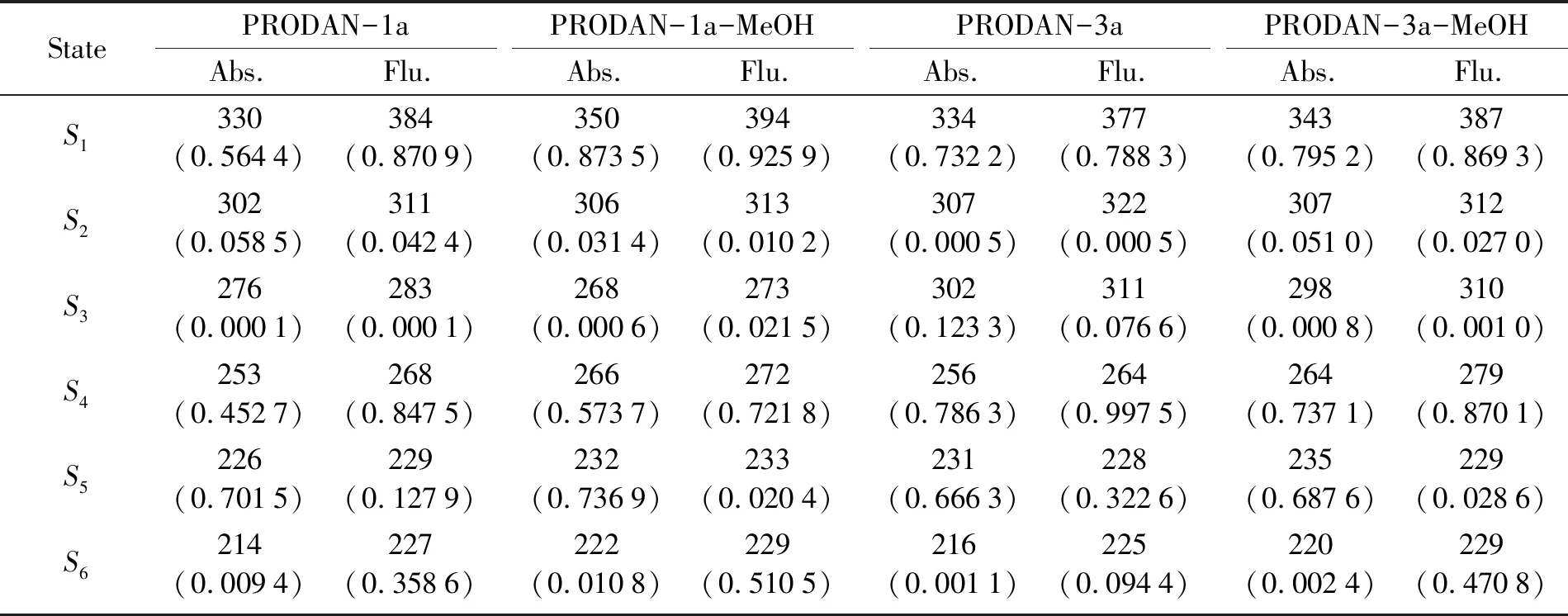
表2 计算得到的PRODAN衍生物(1a和3a)及其氢键复合物的吸收光谱和发射光谱峰值(单位:nm),
从计算的吸收光谱可见(见图4),PRODAN衍生物1a分子在气相中的吸收光谱峰值为330 nm,而氢键复合物PRODAN-1a-MeOH在甲醇溶液中的吸收光谱峰值为350 nm,二者之间发生了20 nm的红移。PRODAN衍生物3a分子在气相中的吸收光谱峰值为334 nm,氢键复合物PRODAN-3a-MeOH在甲醇溶液中的吸收光谱峰值为343 nm,同样发生了红移,但红移程度略小。
通过计算得到的发射光谱可以清楚的看到(图5所示),PRODAN衍生物1a和3a分子单体的发射光谱峰值分别位于384和377 nm;其所形成的氢键复合物的发射光谱峰值依次位于394和387 nm。不难发现,PRODAN衍生物1a和3a分子在甲醇溶液中形成的氢键复合物相对于其气相中的单体,发射光谱峰值也发生了红移,这应该归因于甲醇溶液中分子间氢键作用的增强。
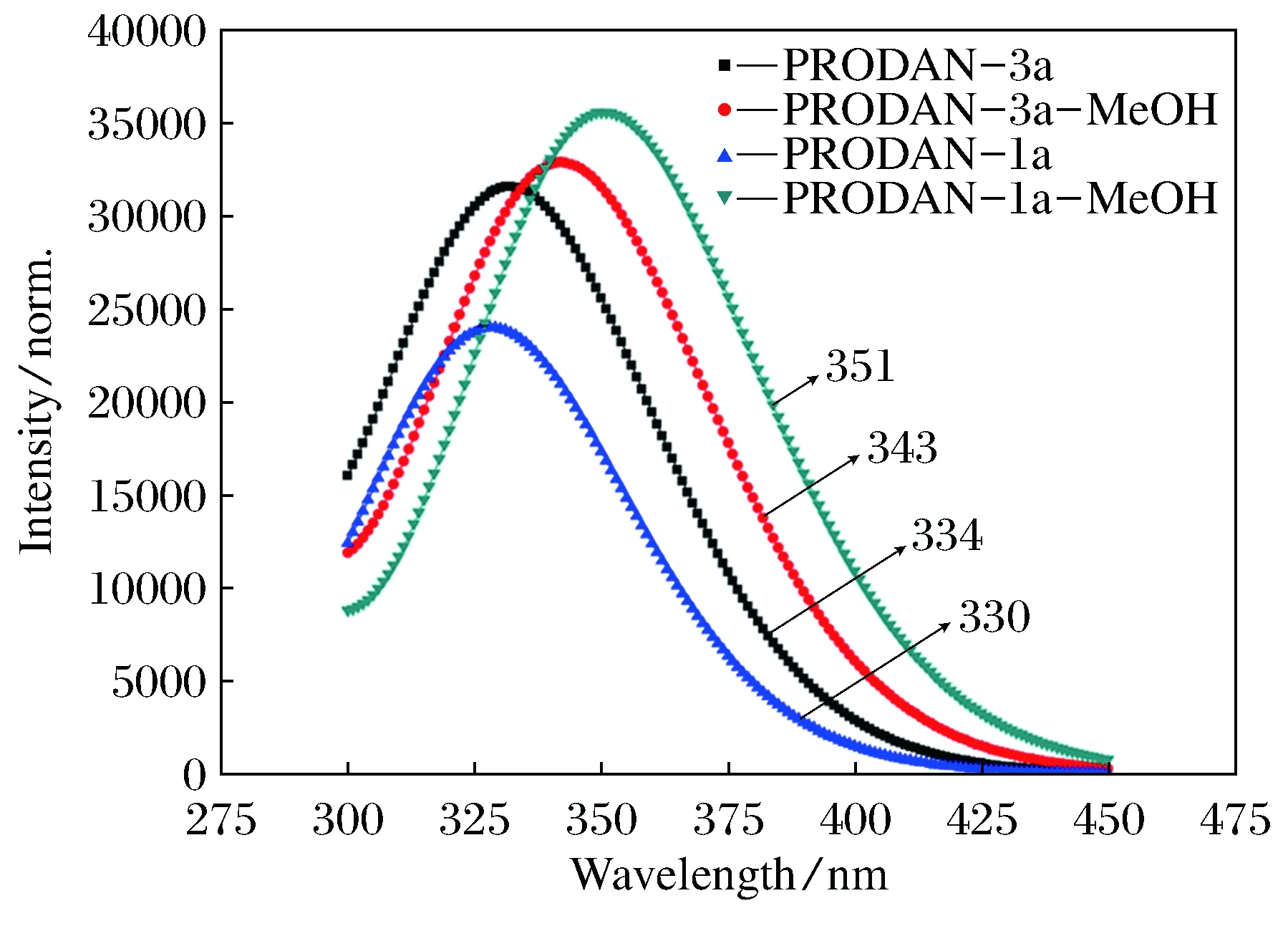
图4 计算的PRODAN衍生物(1a和3a)及其氢键复合物的吸收光谱
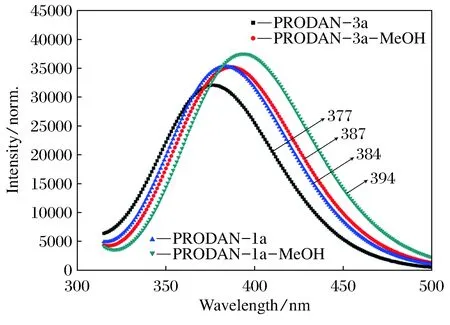
图5 计算的PRODAN衍生物(1a和3a)及其氢键复合物的发射光谱
由计算得到的电子光谱分析,氢键复合物相对单体而言,其吸收光谱和发射光谱都发生了不同程度的红移。其中,1a在溶液中形成氢键复合物后的吸收光谱红移的程度最大,可能在于PRODAN衍生物1a分子中含有分子内氢键。
2.3 分子间氢键结合能
为了更好探究光激发对氢键复合物的分子间氢键作用产生的影响,在TDDFT水平上继续计算了分子间氢键作用结合能。由表3可知,氢键复合物PRODAN-1a-MeOH和PRODAN-3a-MeOH的分子间氢键的总体结合能分别为31.77和32.03 kJ/mol。激发态氢键复合物的分子间氢键的结合能变为40.70和47.22 kJ/mol,表明分子间氢键的强度增强,这和上述对键长和电子光谱的分析所得的结论是一致的。

表3 计算的PRODAN衍生物(1a和3a)及其氢键复合物的总能量(a.u.)和氢键结合能EHB(kJ/mol)
3 结 论
通过DFT/TDDFT/CAM-B3LYP/6-31G**的方法对PRODAN衍生物1a和3a分子分别在气相和甲醇溶液中的激发态氢键对其结构和光谱等性质的影响进行了研究。分析发现,PRODAN衍生物1a和3a分子在甲醇溶液中所形成的氢键复合物的分子间氢键作用明显强于其在气相中的氢键作用。同时,电子光谱显示出甲醇溶液中的氢键复合物吸收光谱和发射光谱都发生了红移;另外,通过对氢键结合能的分析,同样表明在甲醇溶液中的氢键复合物的分子间氢键作用更强。
猜你喜欢
大学物理(2022年9期)2022-09-28
食品工业(2022年3期)2022-03-25
汕头大学学报(自然科学版)(2020年4期)2020-12-14
江苏理工学院学报(2020年2期)2020-10-23
山东化工(2020年15期)2020-09-01
物理通报(2020年7期)2020-07-01
食品安全导刊(2019年18期)2019-09-23
原子与分子物理学报(2015年3期)2015-11-24
原子与分子物理学报(2015年3期)2015-11-24
特产研究(2015年2期)2015-03-24