
7Li2(0,±1)分子体系基态振-转能级的全电子计算*
2019-08-27 00:22王巧霞王玉敏马日闫冰
物理学报 2019年11期
王巧霞 王玉敏 马日 闫冰
(吉林大学,原子与分子物理研究所,吉林省应用原子与分子光谱重点实验室,长春 130012)
1 引 言
Li2是最“轻”的金属同核双原子分子,由于只包含6个电子,Li2成为在理论和实验上研究中性原子间的相互作用的理想模型.因而,很早就在实验上开展了其基态光谱及基态势能曲线的分子轨道理论从头计算研究工作[1−7].早在20世纪70年代,理论上就预测了负分子离子 Li2–1的存在[8],是最小的稳定的负分子离子.作为检验实验与理论的原型分子,中性Li2分子的研究引起了人们广泛的兴趣,对其正负离子体系的研究也逐步开展起来.
中性Li2分子体系的光谱研究开展较早.例如,通过激光诱导荧光和傅里叶变换谱获得了Li2分子激发态-基态跃迁光谱,并得到了电子基态v< 41的部分振动-转动能级结构信息[2].中性分子的理论计算研究也比较充分,之前的文献中采用了各种方案,例如模型势、有效赝势结合极化势、全电子的耦合簇和组态相互作用方法[3,4]等,研究了分子的电子基态与激发态势能函数与光谱常数等.近期的文献综述了中性分子基态的势能函数和振-转能级信息的实验研究; 而与此相比,离子体系的光谱实验研究相对较少.早期的实验[5,7]确定了正离子体系的解离能和基态的部分光谱常数; Sarkas等[9]观测到 Li2–1基态 X2∑u+的负离子光电谱,并通过Franck-Condon分析,获得了体系基态的转动常数Be=(0.502 ± 0.005) cm–1,平衡核间距 (键长)Re=(3.094 ± 0.015) Å和谐振频率we=(232± 35) cm–1.对于离子体系,近期仍有各种计算方法研究电子基态与激发态光谱的文献[10−13]发表,但精确的振-转谱信息研究较少[11].最近的研究[14−17]表明,一些双、三原子分子低电子态振-转谱和分子性质可以通过精确计算获得或重现.
本文通过从头计算研究7Li2(0,±1)分子体系的基态势能曲线,进而得到其光谱常数和振动-转动能级信息.
2 计算方法
本文采用包含单参考耦合簇理论(couple cluster theory with singly,doubly and perturbative triply excitation,CCSD(T)[18−20])和 多参考(multireference,MR)耦合簇理论MRCCSD[21]计算研究了7Li2(0,± 1)分子体系电子基态的势能曲线.计算采用了相关一致基组[22]aug-cc-pwCVXZDK (X=T(3),Q(4),5) (简写为 XZ),在有些情况下,我们在XZ基础上增加了部分弥散基函数;计算中一般关联了体系中所有电子,并通过二阶Douglas-Kroll哈密顿[23]考虑了标量相对论效应.势能曲线计算中,原子核间距从2 Å左右计算到200 Å; 单参考计算中,以 Hartree-Fock 波函数为参考波函数; 多参考计算中,以完全活性空间多组态波函数为参考波函数,活性空间包括Li原子的1s,2s与 2pz 轨道.获得分子体系势能曲线后,通过求解径向薛定谔方程[24]获得分子体系的光谱常数与振-转能级信息.本文中分子的振-转能级写为

其中v为振动量子数,J为转动量子数,Ev,J为径向方程的本征值,G(v)为振动能级;Bv,Dv和Hv等为不同阶次的离心畸变转动常数.
3 结果与讨论
3.1 7Li2分子的光谱常数与振-转能级
图1 为 CCSD(T)/5Z 方法计算的 Li2(0,± 1)分子体系电子基态的势能曲线,能量零点取为各自平衡核间距位置处的能量.表1为7Li2(X1∑g+)分子光谱常数的计算值,并比较了给出的实验值[2,6].首先,采用小基组(TZ)考查了Li的1s电子关联对光谱常数的影响.之前的文献[3]中,完全组态相互作用结合赝势(FCIPP)的计算表明,Li的1s芯轨道的极化效应对光谱常数影响不大; 本文通过比较vCCSD(T)/TZ与CCSD(T)/TZ的全电子计算,结果表明,芯轨道电子关联对光谱常数的影响是不可忽视的,例如对分子平衡核间距Re影响约为0.02 Å,对解离能De和谐振频率we的影响分别为 0.008 eV 和 4 cm–1,对非谐性频率wexe和转动常数Be影响相对较小.表1也研究了光谱常数对基组变化的收敛性.计算结果发现,5Z基组重复了Re的实验值,同时也改进了解离能De数值,缩小与实验值的误差为0.007 eV (误差小于0.7%);振动与转动常数亦随基组增大而逐渐趋于常数,虽与实验值仍小有差距,但考虑到振动、转动常数的拟合与选取的能级数量有关,在下文中给出v=0—24每条振动能级及相应转动常数与实验值的比较(详见表2及下文).

图1 Li2(0,±1) 的基态势能曲线 (能量零点取为各自平衡核间距处能量)Fig.1.Potential energy curves of ground states for Li2(0,±1)(the energy zero-point is located at respective equilibrium internuclear distance).

表1 7Li2(X1∑g+) 分子的光谱常数Table 1.The spectroscopic constants of7Li2(X1∑g+).
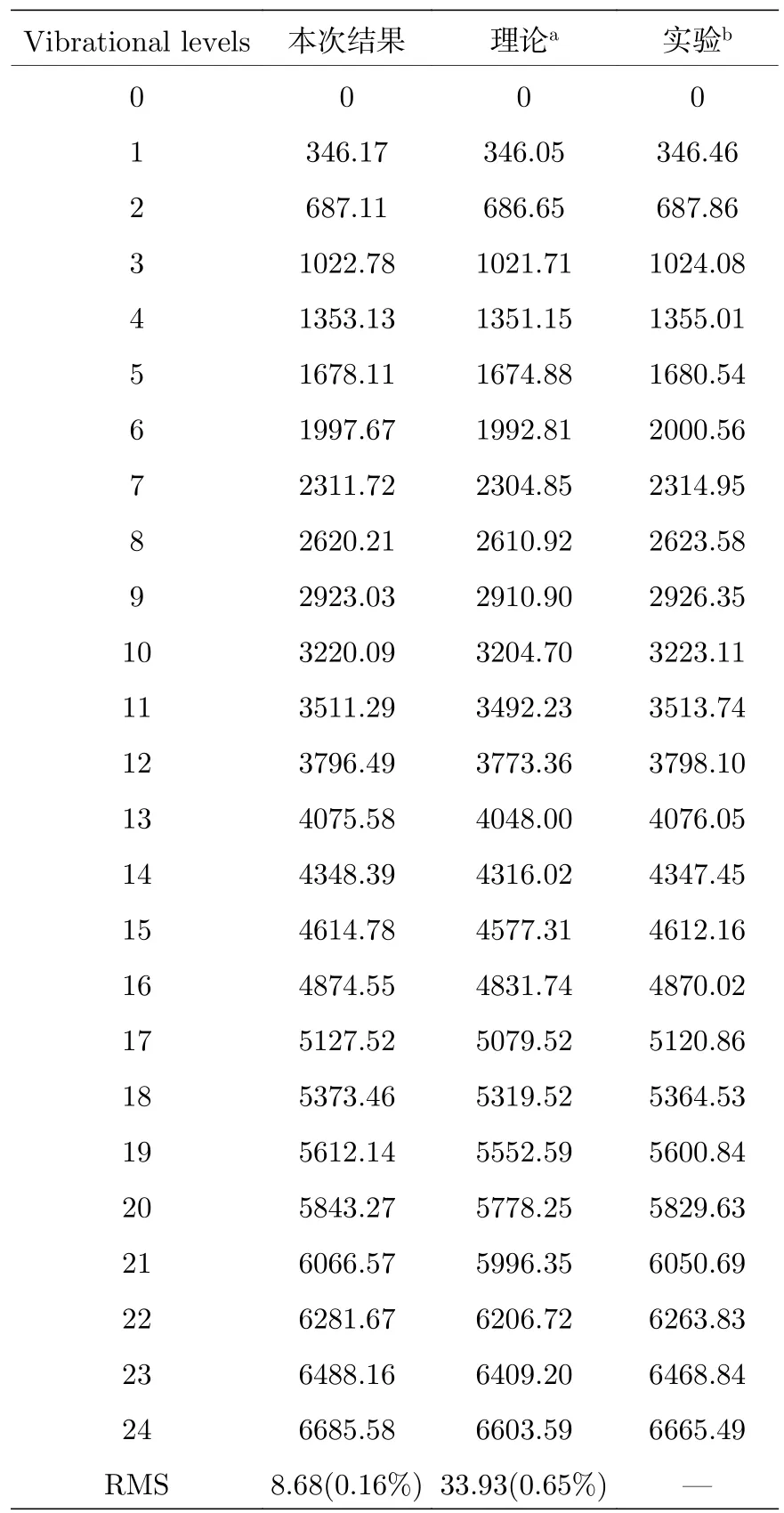
表2 7Li2(X1∑g+)分子的振动能级 Gv (J=0)(单位: cm–1)Table 2.The vibrational levels Gv (J=0) of7Li2(X1∑g+) (unit in cm–1).
表2给出了CCSD(T)/5Z方案计算的分子基态的v=0—24 振动能级,以及前人的理论计算值[3]和实验值[2,6].本文计算的零点振动能为175.006 cm–1,与实验值[6]175.032 cm–1符合得很好.对于最低的16个振动能级,本文计算值与实验值的最大误差约为 3 cm–1; 尤其对于v=8—16 能级,之前的理论值误差为 10—30 cm–1,而本文的计算缩小为几个cm–1.最低的25个振动能级,与实验值相比,本文的均方根误差为 8.68 cm–1(0.16%),较之前的 FCIPP 计算结果 33.93 cm–1(0.65%)明显更优.本文计算结果的改进主要得益于考虑全部6个电子的关联效应,尤其对于长程相互作用,芯电子关联更加重要,而之前的文献中将1s轨道作为赝势处理,对芯-价电子关联考虑不足,因而本文的计算获得的精确振动能级数目更多.总体而言,当前的单参考方法处理Li2中性分子基态是比较成功的.但仍然可以看到相对较高的振动能级 (例如v=23,24),虽然优于前人的计算结果(~60 cm–1),本文的绝对偏差仍达到了约 20 cm–1;这些误差一方面来源于误差的累积,另一方面应来源于非Born-Oppenheimer效应的影响、计算采用了单参考方法等.
表3列出了 Li2分子基态转动常数Bv,Dv(v=0—24振动能级)的本文计算值和已有的实验值[2,6].Bv的计算值与实验值符合得很好,对于低振动态,符合的有效数字达3位; 高阶转动常数Dv处于 10–5cm–1量级,本文计算值随振动量子数v变化趋势与实验测量值一致,低振动态达到两位有效数字符合.接下来,采用类似的计算方案研究正负分子离子体系7Li2±1.

表3 7Li2(X1∑g+) 分子的各振动能级的转动常数Bv与DvTable 3.The rotational constants Bv and Dv of7Li2(X1∑g+).

表4 7Li2±1 分子体系基态的光谱常数Table 4.The spectroscopic constants of ground-state7Li2±1 systems.
3.2 7Li2±1体系的光谱常数与振-转能级
表4给出了计算的分子体系7Li2±1基态的光谱常数,并与一些已有的实验与理论值进行了比较.其 中 Li2+1的基态为2∑g+,Li2–1的基态为2∑u+,计算中分别考虑了其全部的5或7个电子的关联.对于Li2+1基态的Re各种方法计算的结果接近,彼此间差距小于 0.03 Å,其中一种量子蒙特卡罗方案(diffusion quantum Monte-Carlo,DMC)与实验最接近,其他方法与实验值相比,误差为0.01—0.02 Å.文献中各种方法计算的 Li2+1基态解离能De与实验值相比至少有三位有效数字相互吻合,本文RCCSD(T)/5Z的计算结果与实验值有四位有效数字吻合,误差小于 3 cm–1.为了检验这一开壳层体系波函数的多组态性质,本文中还采用了多参考方法MRCCSD计算了势能曲线,由于方法极其耗时,我们仅使用了TZ级别的基组.计算表明,在多参考方法下,芯电子关联的影响仍不可忽视,对Re,we,和De的影响分别约为 0.03 Å,4 cm–1和 0.015 eV.本文采用的多参考方法对键长略有改进,进而使转动常数也更贴近实验值; 解离能的变化为 0.003 eV (~24 cm–1).各种方法计算的振动、转动常数均与实验值合理吻合.同时,我们注意到之前文献中的CI方法也给出了与本文单参考、多参考方法相近的计算结果,可见基态Li2+1分子离子波函数的多组态性质不明显,单参考理论对其描述也是合理的.
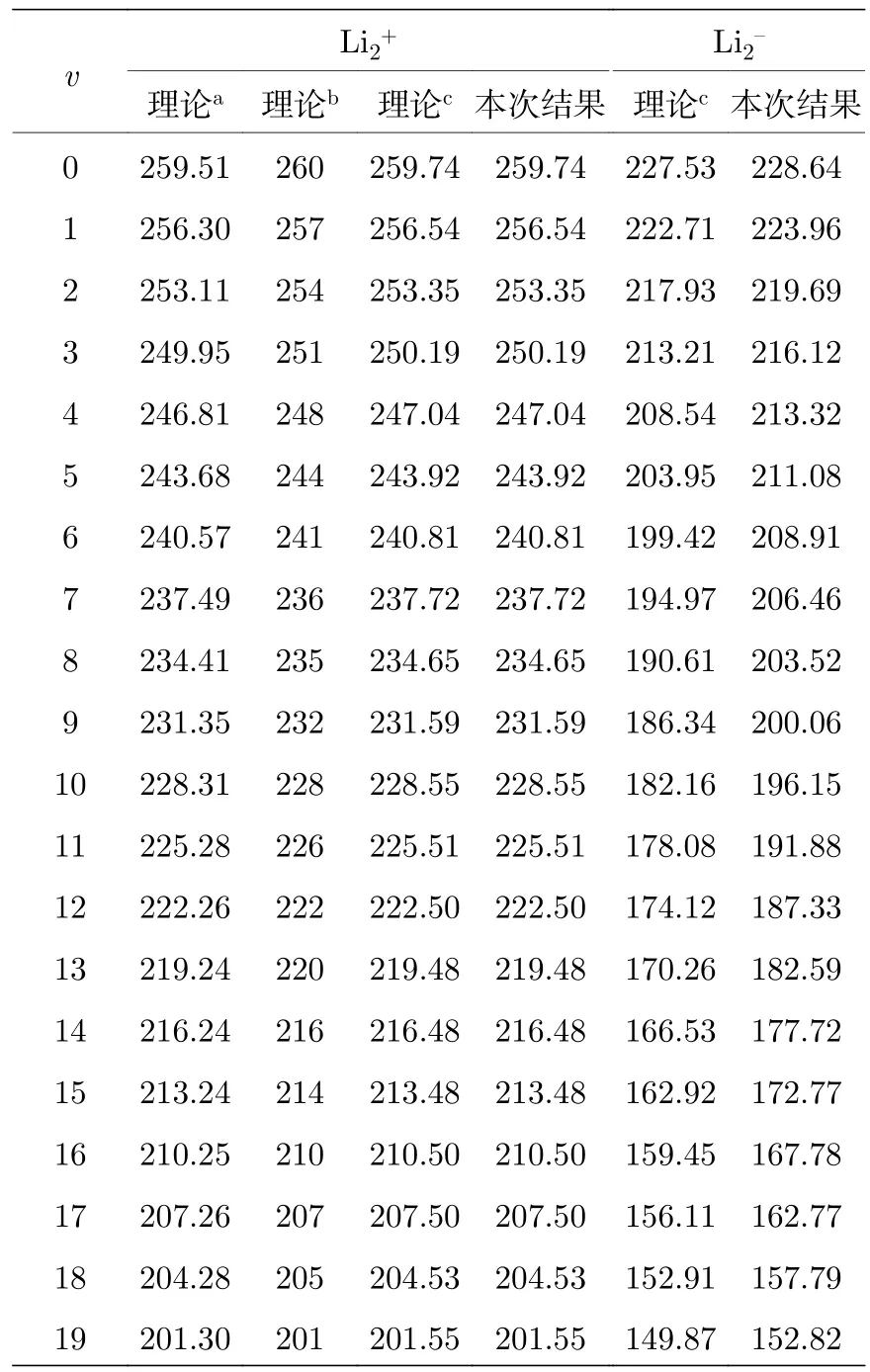
表5 Li2± 基态振动能级间隔 G (v + 1)–G (v)(单位: cm–1)Table 5.The vibration energy spacing G (v +1)–G (v) of ground-state Li2± (unit in cm–1)).
对于负离子体系的Re和De,各计算方法的结果彼此间差异较大.对于 Li2–1的Re,彼此之间相差达0.1 Å,其中DMC方案给出了最接近实验的结果; 但是,对于解离能De,该方法的计算误差最大 (接近 0.1 eV); 也注意到,DMC 方法给出了最贴近实验值的Re,但同时却给出了偏离最大的Be,略显不自洽.为了更好地描述负离子体系,我们在5Z的基础上,增加了4个s型和2个p型弥散高斯基函数,即采用 CCSD(T)/5z + 4s + 2p 计算了Re,发现数值改进了约 0.2 Å,但由于存在收敛性等技术问题,没有得到完整的势能曲线以获得光谱常数.之前文献中MRDCI方法获得的计算结果[11]的与实验值接近 (误差~0.03 Å),可见该负离子体系的波函数多组态性质可能更为明显.因此我们认为对此体系结果的改进可通过增加弥散基和采用多组态或多参考方法实行.为此,本文中采用了 MRCCSD/TZ + 4s + 2p 方案也计算了基态势能曲线并给出了光谱常数(表4); 由于计算是非常昂贵的,一方面采用了小基组,另一方面限制了活性空间大小,除了1s2s轨道仅包含了额外的2pz轨道.由表4中可见,多参考方法对键长确有改进(0.013 Å),但与实验值仍有差距.我们认为由于本文已经采用了足够的弥散基函数,这一差距主要来源于多组态展开中包含的s轨道数目不足.在表5 中给出了7Li2± 1分子离子基态的v=0—19的振动能级间隔,结果表明,各方法计算的振动能级间隔相互符合得很好,误差为几个cm–1量级.可见,各方法对振动能级的计算应是可靠的.为此,本文中还给出了相应的转动常数Bv,Dv,见表6;虽然v=0的结果B0与实验给出的Be合理接近,目前仍缺少详细的实验结果可供比较.还需指出的是,其他的 Li20,±1同位素分子体系的部分光谱常数,如we,Be等可由同位素关系直接得到; 并且由于获得了体系的势能曲线,上述体系的同位素分子的振-转能级信息也可以通过求解径向方程获得,为方便以后的理论与实验比较,表7中直接列出了中性体系7Li2的两种同位素分子的振-转能级信息.

表6 7Li2± 基态分子的各振动能级的转动常数Bv与DvTable 6.The vibrational levels Bv and Dv of7Li2±.

表7 Li2分子的同位素体系的振动能级与转动常数Table 7.The vibrational levels and rotational constants for isotope molecules of Li2.
4 结 论
采用CCSD(T)与 MRCCSD方法计算了7Li2(0,±1)分子体系的基态的势能曲线,并以中性分子为例讨论了基组效应及芯-价电子关联的影响.CCSD(T)/5Z计算的势能曲线获得了与实验吻合很好的光谱常数,进而计算了7Li2的振动-转动能级,计算的振动能级较之前的FCI结合赝势的计算结果有较明显的改进.对于阳离子分子体系,以单参考与多参考耦合簇理论计算了势能曲线并拟合了光谱常数,二者计算得到的光谱常数十分接近,从而证明了采用单参考组态波函数描述阳离子分子体系基态波函数是合理可行的.对于负离子分子体系,单参考理论计算获得的平衡核间距与实验值存在差距,已经讨论了误差来源,并通过增加弥散高斯函数与采用多参考理论缩小了与实验值的差距; 其他光谱参数与实验值符合较好.对于上述分子体系,文中均给出了振动能级信息和转动能级参数.通过当前的精确计算研究,可加深对上述分子体系电子结构与光谱的认识.
猜你喜欢
无线电工程(2022年10期)2022-10-24
——《势能》
文化纵横(2022年3期)2022-09-07
当代党员(2022年9期)2022-05-20
延边大学学报(自然科学版)(2021年1期)2021-04-27
华人时刊(2021年23期)2021-03-08
复旦学报(医学版)(2020年3期)2020-06-18
高中生学习·高三版(2017年9期)2017-10-26
中学生数理化·高三版(2017年1期)2017-04-20
中学生数理化·八年级物理人教版(2016年5期)2016-08-26
新高考·高一物理(2015年3期)2015-08-20
