
共轭聚合物内非均匀场驱动的超快激子输运的动力学研究*
2019-09-21 05:56王文静李冲张毛毛高琨
物理学报 2019年17期
王文静 李冲 张毛毛 高琨‡
1) (齐鲁师范学院物理与电子工程学院,济南 250013)
2) (山东大学物理学院,晶体材料国家重点实验室,济南 250100)
1 引 言
基于共轭聚合物的有机太阳能电池 (organic solar cell,OSC),因其质量轻、柔性半透明、光电特性易调制以及可大面积卷对卷印刷等优势,近年来备受关注,并成为目前热门的研究领域之一[1-3].随着新型共轭聚合物等有机分子的合成、器件结构的优化和光伏机理认识的不断突破,单结有机太阳能电池的光电转换效率已突破15%[4],利用多结器件的互补吸收,其级联器件的效率甚至已达到了17.3%[5].尽管如此,与近年同样备受关注的有机/无机杂化钙钛矿太阳能电池和传统的硅基太阳能电池相比,有机太阳能电池的效率仍然偏低.特别是,有机太阳能电池的效率距其理论预测的极限值存在不小的差距[6].因此,如何在大面积的光伏模块上实现稳定的高效率值依然是有机太阳能电池研究急需解决的问题[7].造成这一现状的根本原因是聚合物等有机分子较强的电子-晶格相互作用.由于该作用,聚合物内光激发产生的电子-空穴对同时诱导晶格畸变,使其被束缚在晶格势场中,最终形成空间局域的激子[8-10],其局域度可以通过引入束缚能定量描述.据报道,聚合物内的激子束缚能约可达到 0.5 eV[11,12],仅靠室温热能(0.026 eV)无法将其解离为自由载流子形成光电流.实际应用中,一般是把聚合物光伏体系设计为电子给体(D)/受体(A)结构.当光激发产生的激子输运至D/A界面,便可在界面势差异诱导的驱动力作用下实现解离,此即为由激子图像主导的电荷分离过程[13,14].
同时,由于聚合物内的激子可通过辐射或非辐射跃迁的方式退激发至基态,时间尺度一般为100 ps-1 ns,即激子寿命[15].因此,激子图像主导的电荷分离过程要求激子必须在其寿命之内输运至D/A 界面,否则,光激发的能量将会被耗散掉.目前,聚合物等有机体系内的激子输运一般被认为是由Förster[16]和Dexter[17]两种机制决定的激子扩散过程.由于扩散速度的限制,激子在聚合物内的扩散长度约仅能达到10 nm[18,19],这远小于聚合物的光吸收长度(约100 nm).因此,早期的双层平面D/A异质结有机太阳能电池要求功能层非常薄,这虽然提高了激子到达D/A界面的效率,却同时降低了光的吸收效率,导致器件光伏性能并没有达到预期.基于此,研究人员进一步改进了有机功能层的结构,设计了D/A分子共混的体异质结太阳能电池,使上述问题得到解决,并由此大幅度提高了器件光伏效率[20].但是,基于D/A体异质结构的光伏层内,由于激子扩散长度的限制,D/A相分离尺度要控制在10 nm范围以内,这对于聚合物等柔性的有机体系难以完全实现.特别是,如此小的相分离尺度大幅度限制了载流子在光伏层内的输运,这对器件光伏性能的进一步提升造成了困难.可见,要设计更高效的有机太阳能电池,必须对激子输运机制有新的认识,并在此基础上探索实现激子超快输运的方法.
聚合物体系内的激子是否可以实现超快输运呢? 随着飞秒超快技术和瞬态探测技术的发展,一些最新的实验现象间接或直接证实了这一疑问.2012年,Kaake等[21]率先通过瞬态光致吸收光谱实验指出,在某些高效的聚合物光伏体系内,有高达70%的电荷可在100 fs内实现分离.根据由Förster和Dexter两种机制决定的激子扩散过程,激子不可能在如此短的时间内集中输运至D/A界面实现电荷分离.那么,是什么原因导致了聚合物光伏体系内如此高比例的超快电荷分离呢? 目前,主流的图像把原因归结为激子在形成之初的离域性[22-24],即在聚合物分子内初始的光激发态在前100 fs内并未完成弛豫.激发态的离域性一方面有效地降低了电子-空穴对的束缚能,另一方面大幅度地提高了其输运速度.激子超快输运更直接的实验证据也相继被报道[25,26].比如,Jin等[26]于2018年设计了一种有机纳米纤维,由poly(di-nhexylfluorene)分子晶状物作核,polyethylene glycol和polythiophene作为冠状分子分别与核的中间和两端相连.这种非均匀的构型由于存在激子产生能的梯度,导致激子沿着核由中心位置向末端位置超快输运,输运长度达到200 nm.总体而言,人们已经认识到激子超快输运的实现取决于聚合物体系的构型及微观形貌,但是,对于激子超快输运的机制和更详细的动力学过程仍缺乏足够的认识.基于以上问题,本文提出了聚合物内由非均匀场驱动的超快激子输运机制,并对其输运的动力学过程进行了模拟和讨论.
2 模型与方法
共轭聚合物一般由sp2杂化的碳原子聚合而成,如聚乙炔 (PA)、聚对苯乙炔(PPV)和聚噻吩(PT)等,由于其2pz轨道中的π电子未参与成键,可沿分子链方向发生离域,导致此类聚合物具备功能性.本工作为简单起见,选取具有基态非简并特性的顺式聚乙炔(cis-PA)作为研究对象.为了突出该体系较强的电子-晶格相互作用和准一维的特点,采用扩展的Su-Schrieffer-Heeger (SSH)模型[27].根据顺式聚乙炔分子的链状结构,其哈密顿量为(忽略电子自旋指标)
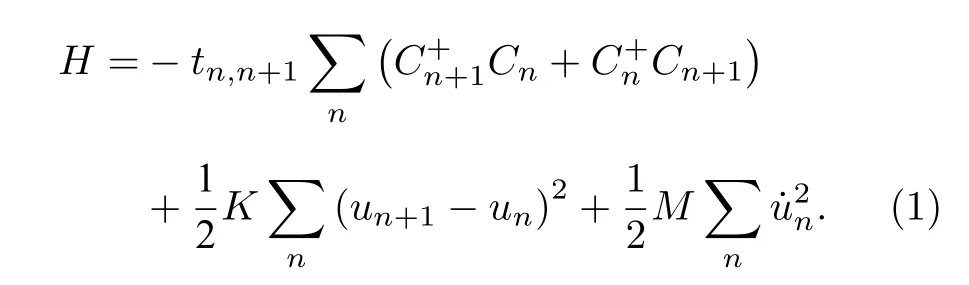
其中,第一项描述了电子在相邻格点n和n+ 1之间的跃迁,
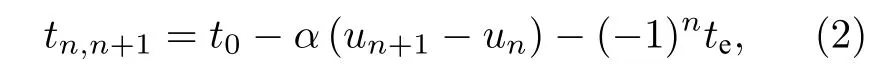
t0为格点均匀排列时相邻格点之间电子的跃迁积分,α为电子-晶格相互作用常数,un为第n个格点相对其平衡位置的位移,te则突出了顺式聚乙炔的基态非简并特性,称为简并破缺参数.为第n个格点上的电子产生(湮灭)算符.(1)式中的第二项和第三项分别从经典的角度描述了分子链格点的弹性能和动能.K为格点间弹性力常数,M为格点质量.
由以上哈密顿量出发,可以求解体系的晶格结构和电子结构.当体系处于基态时,其晶格结构呈现均匀二聚化的特点,而其电子结构则具有半导体的性质[9,11].如果体系受激,价带内的电子获得能量可以跃迁至导带,形成激发态.考虑带边跃迁,价带顶的一个电子跃迁至导带底,晶格完全弛豫后则形成激子[10].本文的理论模拟将以激子作为初始态,考虑它在非均匀场下的动力学演化过程 (不同场的哈密顿量将在下节分别给出).无外场时,体系保持初始态不变,一旦施加外场,体系将经历演化.晶格的演化遵从经典的牛顿运动方程
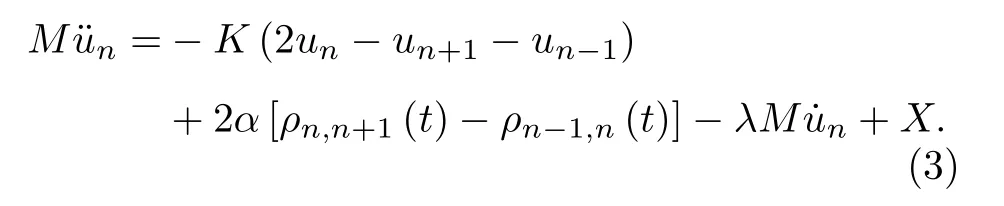
其中,ρn,n′(n′=n±1) 为密度矩阵,定义为
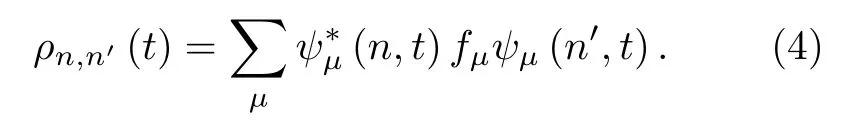
ψµ(n,t)=〈n|ψµ(t)〉是电子态 |ψµ(t)〉 在Wannier表象中向 |n〉 的投影.fµ(=0,1,2)是与时间无关的电子分布函数,仅由初始态的电子占据决定.另外,为了描述格点能量向外界环境中的耗散,(3)式中引入了格点振动的阻尼项,λ为阻尼因子.体系ψµ(n,t)的演化则遵从含时薛定谔方程

需要强调的是,(3)式和(5)式中的X和Π分别为外场对体系晶格和电子态演化的影响,具体形式将在第 3节根据不同的外场给出.从(3)式和(5)式可以看到,体系晶格和电子态在整个演化过程中是耦合在一起的.这说明晶格的演化可以在不同势能面上展开,即本工作采用的是非绝热的量子动力学方法.固定边界条件下,耦合方程(3)式和(5)式通过8阶可控步长的Runge-Kutta方法求解.
3 结果与讨论
模型中的参数根据顺式聚乙炔选取[28]:α=41 eV/nm,t0=2.5 eV,te=0.05 eV,K=2100 eV/nm2,M=1.35×106eV·fs2/nm2.尽管如此,本文的结论同样适用于其他聚合物分子体系.下面,本文将结合聚合物光伏体系內禀存在或外界可调的两种非均匀场(包括由极化或受限电荷诱导的非均匀电场和分子排列相关的非均匀构型场),分别提出它们的理论模型,继而对它们驱动的激子超快输运动力学过程进行理论模拟.在动力学模拟之初,假定激子已在聚合物链中产生.
3.1 非均匀电场驱动的激子超快输运
在实际的聚合物光伏体系内,非均匀电场总是內禀存在的.在其D/A界面附近,聚合物链的取向更为无序,各种缺陷也不可避免,这造成D/A界面附近的部分电荷被限制,从而形成受限电荷.另外,由于D/A分子结构差异及它们的相对位置和取向不同,D/A界面附近电荷密度存在突变.这些因素都会诱导D/A界面附近存在局域的非均匀电场[29,30].除了内禀因素外,通过向聚合物光伏体系掺杂向电性添加剂(electrotropic additives)或具备极性的分子,其内部的非均匀电场也可以人为的产生或调控[31,32].以往工作中,通过引入线性梯度的电场已经对激子的超快输运和解离动力学过程进行了模拟,并基于此解释了有机太阳能电池内观察到的超快电荷分离现象[33].本工作选取更接近实际情况的非均匀电场形式.假定聚合物链的右端点附近存在一个受限正电荷(电荷量为 |e|),如图1所示,d为电荷与链右端点的距离.根据点电荷的电场形式,受限电荷诱导的电场沿聚合物链的分布为
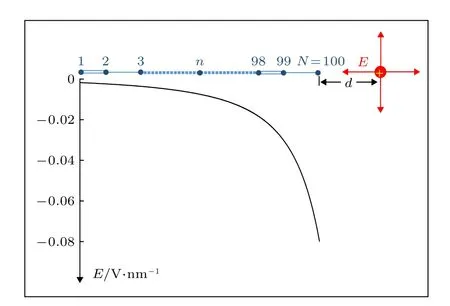
图1 受限正电荷 (电荷量为 |e|)相对于聚合物链的位置,d为电荷与链右端点的距离; 曲线为d=3 nm时电场强度沿分子链的分布,负号表示电场的方向与分子链的正方向相反Fig.1.Schematic diagram about the position of a confined charge (q=|e|) relative to the polymer chain,d shows the distance between the charge and the right chain-end; the curve describes the distribution of the induced electric field E along the polymer chain with the case of d=3 nm,where the minus sign means that the direction of the electric field is opposite to that of the chain.
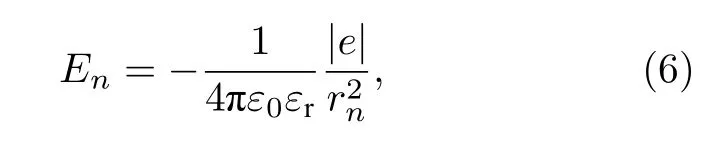
其中,负号表示电场的方向与分子链的正方向相反,ε0为真空介电常数,εr(=2)为聚合物链的相对介电常数.rn=[(N-n)a+un+d]是聚合物链第n个格点至受限电荷的距离,N=100为链总格点数,a=0.122 nm为链晶格常数.
在动力学模拟中,为了避免数值错误,电场En并不是瞬时开启的,而是以半高斯的形式平滑地随时间t开启,即
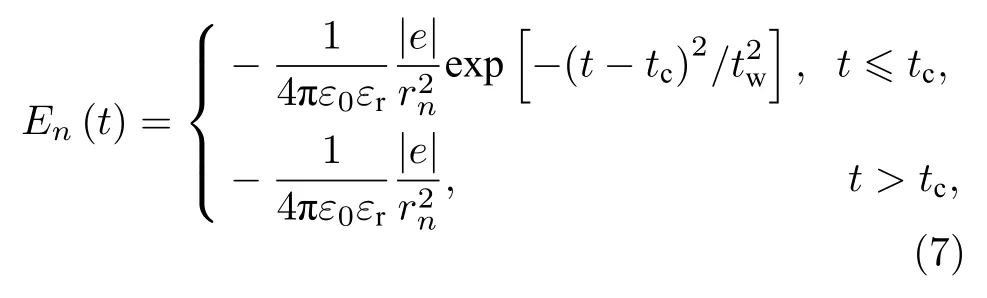
其中,tc为电场达到稳定分布所对应的时刻,tw是电场开启过程相关的时间参量.通过比较,发现这两个时间参量会对动力学过程产生影响,但本工作突出的是稳定后的非均匀电场对激子输运的影响.因此,以下模拟中这两个时间参量取固定值,即tc=30 fs和tw=15 fs.由此出发,受限电荷诱导的非均匀电场的哈密顿量为

将(8)式与(1)式合并,即可分别得到非均匀电场对体系晶格和电子态演化的贡献,
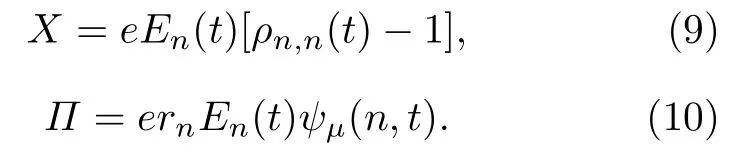
在本工作的模型中,影响非均匀电场在聚合物链上分布的主要因素是受限电荷与链右端点的距离d.图1中的曲线给出了d=3 nm时,开启后(t>tc)的电场沿分子链的分布.可见,电场强度沿分子链的方向迅速增加,呈现出了明显的非均匀特点.以产生在nc=30 (nc为激子中心位置)附近的激子为初始态,一旦开启以上形式的非均匀电场,激子将经历演化.如图2所示,本文给出了激子在演化过程中分子链晶格结构随时间的变化,即yn=(-1)n(2un-un+1-un-1)/4.可以发现,在非均匀电场的驱动下,激子自其初始位置沿分子链向右输运,大约在t=2000 fs (或2 ps)时,激子到达分子链的右端并反弹.就其总体过程而言,激子在2 ps时间内输运的距离达到了5 nm,即其平均输运速度高达2.5 nm/ps.这一速度值比由Förster或Dexter扩散机制主导的激子输运速度值(约0.1 nm/ps)提高了1个量级[16-19].可见,非均匀电场能够驱动激子沿分子链超快输运.
然而,激子作为中性态为什么能够在电场的驱动下实现输运呢? 以往的研究表明[34,35]: 激子在均匀电场的作用下一般呈现极化的行为,即激子内的电子和空穴在电场力的作用下不再完全重合,而是沿分子链有一定程度的分离,从而造成激子内出现等量异号的极化电荷.图3的插图给出了t=1000 fs时激子在电场下的极化电荷分布qn(t)=e[ρn,n(t)-1].可见,激子中心的右侧出现了极化的负电荷,而其左侧则出现了极化的正电荷.如果电场是均匀场,正负极化电荷所处的电场强度相同,那么激子整体所受的电场力为零,激子无法输运.但是,本工作中所采用的电场为非均匀场,极化的负电荷所处的电场强度要高于正电荷,这将导致激子整体受到沿分子链向右∑的电场力,这正是激子输运驱动力的来源,即F=nqn(t)En(t).随着激子沿分子链向右输运,它感受到的电场强度迅速增加(如图1曲线所示),这造成激子的极化程度显著增强.一方面,激子内极化的正负电荷量增多; 另一方面,正负电荷所处的电场差异增强.因此,驱动力F随着时间或激子的输运将迅速增大.定量的计算结果如图3所示,与以上的分析一致.
此处,聚合物链内激子的输运速度可以通过调控非均匀电场沿分子链的分布进一步提高.由(6)式可知,受限电荷与链右端点的距离d是影响非均匀电场分布的重要因素.图4给出了激子的中心位置nc在不同非均匀电场(通过改变d调控)驱动下随时间的演化.很明显,随着距离d的减小,激子将更快地到达分子链的右端.当d=1 nm时,激子中心大约经历1.5 ps就从其初始位置nc=30到达链的右端,并且其中心位置可以达到nc=80,随后激子将在链端反射呈现振荡的行为.可见,当受限电荷与链右端点的距离d减小至1 nm时,激子的平均输运速度可增至约4 nm/ps.至于不同d值时激子到达链端所呈现出的不同振荡行为,这主要取决于不同情况下的激子输运速度.激子输运速度越大,激子的振荡行为越明显.当d=3 nm或d=5 nm时,激子到达链端也会振荡,但振荡周期和幅度将分别变长和变弱.另外,需要补充两点: 一方面,受限电荷与链右端点的距离d存在临界值,当d小于此临界值时,受限电荷在分子链右侧区域诱导的电场将足以使激子解离,相关图像以往已有报道[34,35],此处不再赘述; 另一方面,在模型处理中,本工作假定受限电荷正好处于分子链的延长线上,这与实际情况可能不符.当受限电荷处于任意位置时,对激子输运起作用的电场实际为受限电荷诱导的电场沿分子链的分量.这种情况下,电场分量沿分子链的分布也是非均匀的,以上图像同样适用.
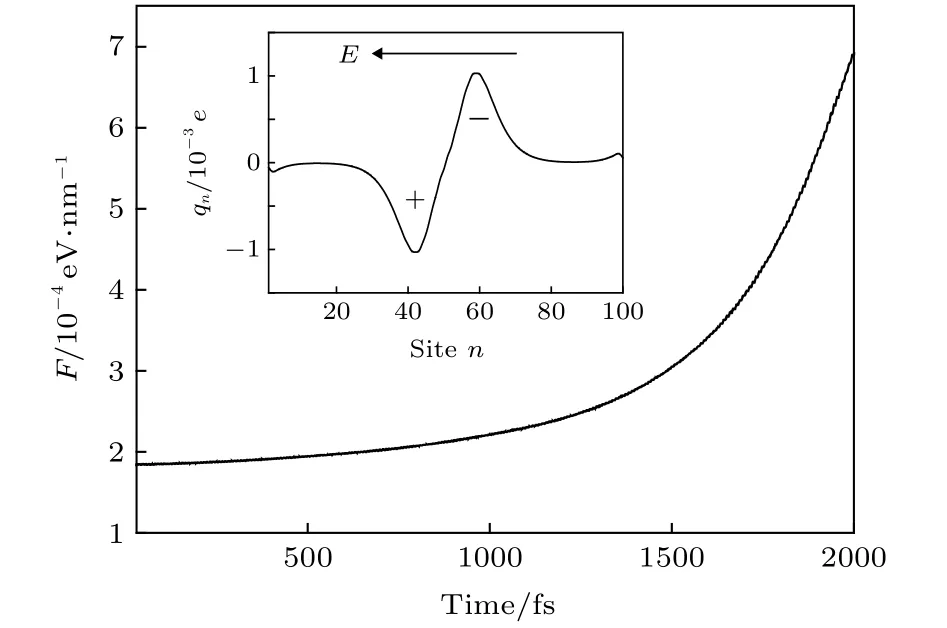
图3 非均匀电场诱导激子输运的驱动力F随时间的变化; 插图为t=1000 fs时激子内极化的正电荷与负电荷的分布Fig.3.Variation of the driving force F as a function of the time.The inset presents the polarized positive charges and negative charges in the exciton at the time t=1000 fs.
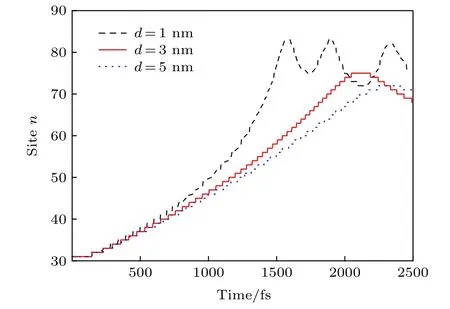
图4 不同非均匀电场(通过改变d调控)驱动下激子中心位置nc随时间的演化Fig.4.Time evolution of the exciton center nc along the polymer chain driven by different nonuniform electric fields,which can be modulated by changing the value of d.
3.2 非均匀构型场驱动的激子超快输运
基于D/A结构的聚合物光伏体系内,特别是D/A界面处,由于侧链基团、空间结构及其他无序效应的影响,聚合物分子之间通常会呈现非均匀的排列[36].比如考虑两条聚合物分子链,在其耦合区域,非均匀的排列将导致它们之间的相互作用也是非均匀的.因此,沿分子链方向必定存在与分子间的排列构型相对应的非均匀场,本文称之为非均匀构型场.为简单起见,本工作构造了两条呈线性排列的分子链,如图5所示.每条分子链的总格点数均为N=100,沿分子链方向垂直最近邻格点间的距离dn呈线性减小的趋势,即
其中,k代表分子链之间线性排列的斜率;a=0.122 nm为晶格常数;dN为分子链右端点对应的垂直最近邻格点间距离,取dN=0.24 nm.引入垂直最近邻格点间的电子跃迁项t⊥(n) 描述分子链之间的相互作用,它的大小取决于dn,见(12)式[37],
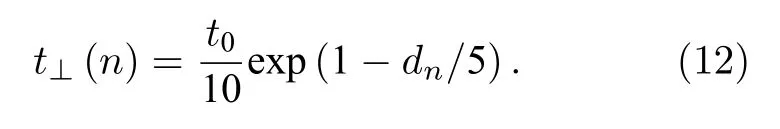
为了研究该非均匀构型场对激子输运的影响,在动力学模拟之初,假定激子已经在第1条链上产生,其中心位置为nc=30.同时,为了避免数值错误,两条分子链之间的相互作用t⊥(n) 也不是瞬时开启的,而是以半高斯的形式平滑地随时间t开启,即
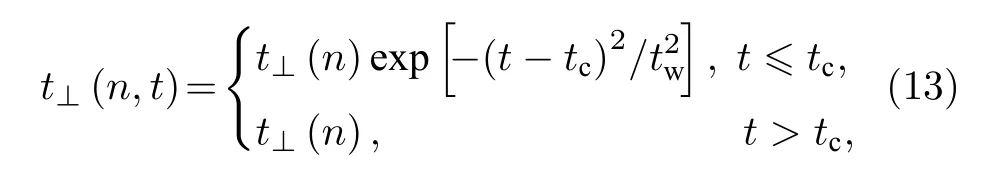
其中的时间参量也固定为tc=30 fs和tw=15 fs.由此出发,分子链线性排列对应的非均匀构型场的哈密顿量为
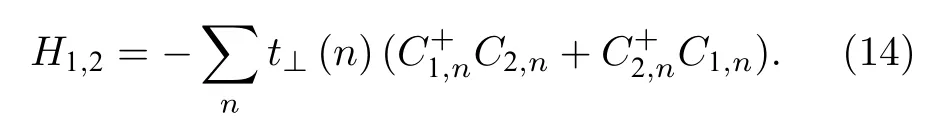
其中1和2分别为两条分子链的指标,如图5所示.将(14)式与(1)式合并,将分别得到非均匀构型场对体系晶格和电子态演化的贡献,
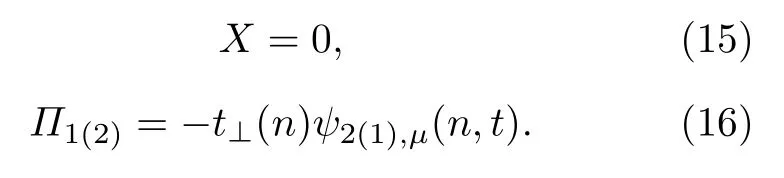
开启以上形式的非均匀构型场后,激子将经历演化.在以上模型中,分子排列的非均匀构型场可通过改变分子链之间线性排列的斜率k进行调控.图6给出了k=0.03时激子在演化过程中分子链晶格结构yn随时间的变化.发现: 在构型场开启之初(1-50 fs),初始产生在第1条链上的激子由于链间相互作用迅速向第2条链扩展,但保持其中心位置nc=30不变; 当激子在两条链之间扩展至一定程度,在非均匀构型场的驱动下,激子自其初始位置沿分子链向右输运,大约在t=200 fs (或0.2 ps)时,激子到达分子链的右端.总体而言,激子在0.2 ps时间内输运的距离达到了约6 nm,即其平均输运速度高达30 nm/ps.这一速度值不仅比Förster或Dexter机制主导的激子输运速度提高了2个量级,也比由前面非均匀电场驱动的激子输运速度提高了1个量级.
下面将深入分析激子在此线性构型场驱动下超快输运的物理机制.以往的静态研究已表明分子链间的相互作用强度将会对激子的产生能有重要影响[12].具体来说,激子的产生能会随着分子链间相互作用强度的增强而减小.本文的模型处理中,分子链间的相互作用强度t⊥(n) 是通过改变分子链间的距离dn来调控的.图7中的插图给出了k=0.03时,激子的产生能 ΔE随分子链间距离dn的变化.很明显,链间距越小,激子产生能越低,这一能量梯度必将沿分子链方向诱导激子输运的驱动力F,可以简单地通过F=-(ΔEn+1-ΔEn)/a求出.如图7所示,本文给出了k=0.03时,线性构型场所诱导的激子驱动力沿分子链的分布.一方面,驱动力为正,这将驱动激子沿分子链正方向输运.另一方面,驱动力沿着分子链的正方向逐渐增强,这将导致激子沿分子链输运的速度逐渐加快,这与图6的模拟结果是一致的.特别是,通过对比非均匀电场和构型场诱导的激子输运驱动力(图3和图7),发现线性构型场诱导的驱动力要比前者高2个数量级,这导致线性构型场驱动的激子输运速度更快.
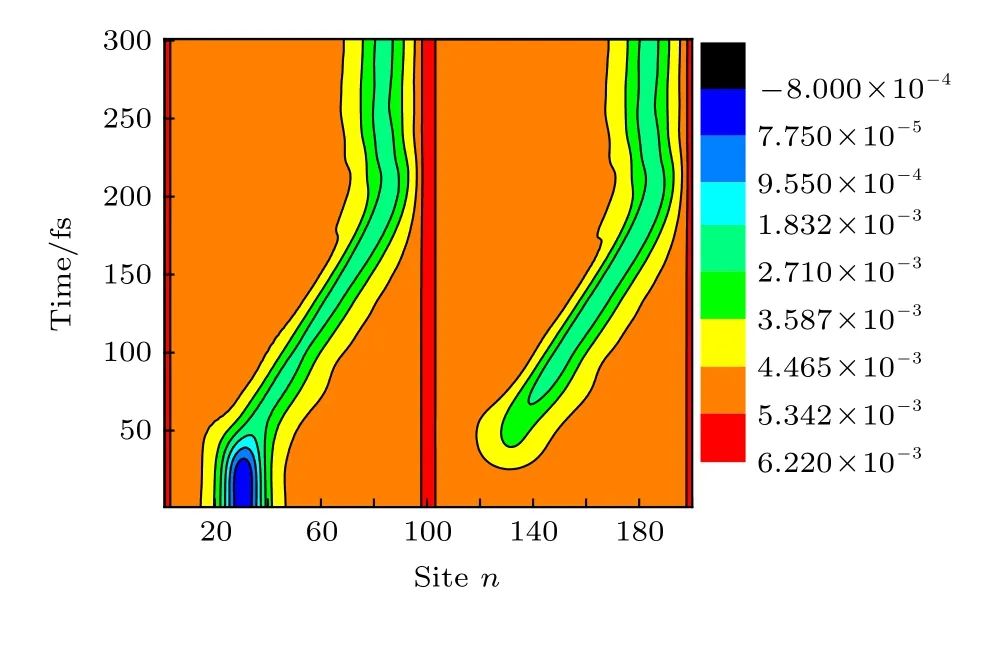
图6 激子在线性链间构型场 (k=0.03)驱动下在分子间扩展及沿分子链输运的晶格动力学演化,激子初始产生在第1条分子链上,中心位置为nc=30Fig.6.Time evolution about the lattice configuration of an exciton in two coupled polymer chains driven by the nonuniform configuration field with a linear coefficient k=0.03.
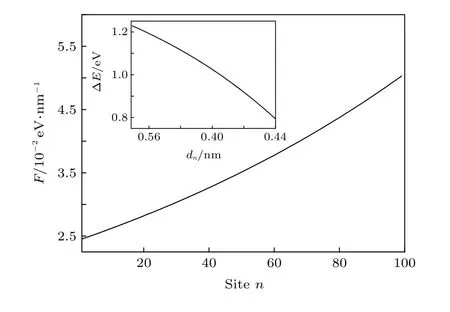
图7 线性构型场(k=0.03)诱导的激子驱动力F沿分子链的分布,插图为激子产生能 ΔE 随链间距离dn的变化Fig.7.Distribution of the driving force F along polymer chains driven by a linear configuration with k=0.03.The inset presents the dependence of the exciton creation energy ΔEupon the interchain distance dn.
根据前面的模型处理 (见(11)式),耦合分子链线性构型场的分布可以通过改变其斜率k来调控,也就是说,激子沿分子链输运的速度必定与k的取值密切相关.图8给出了不同k值时激子中心位置nc随时间的演化.发现: 线性构型场驱动的激子输运速度可以通过改变其斜率k调控.特别是,存在最优化的斜率值k=0.03,此时激子输运速度最快.当k<0.03时,由于沿分子链方向激子产生能的梯度趋于减小,驱动力F必将减弱,因此激子输运的速度将减慢.极端的情况是分子链均匀排列,即k=0时,激子的产生能在分子链各处均相等,激子将保持不动.当k> 0.03时,由于模型处理中固定了链右端点的距离dN=0.24 nm,这导致分子链左侧区域的链间距过大,这对激子产生能的影响将变小,因此激子输运的速度也趋于减慢.同样,可以考虑极端情况,即k→∞时,模型体系左侧分子链间的距离趋于无限远,第2条链对第1条链内产生的激子无任何影响,激子也将保持其初始位置不动.当然,实际的分子排列构型一般不是线性的,形式可能要复杂得多.但是,对于耦合的分子链,其耦合区域的分子间距离总是存在局部的递减趋势,这一变化必将诱导激子产生能沿分子链方向的梯度(或激子输运的驱动力),从而驱动激子超快输运.

图8 不同线性分子排列构型场(通过改变k调控)下激子中心位置nc随时间的演化Fig.8.Time evolution of the exciton center nc along polymer chains driven by different configuration fields,which can be modulated by changing the value of k.
4 结 论
本文结合聚合物光伏体系內禀存在或外界可调的两种非均匀场(包括由极化或受限电荷诱导的非均匀电场和分子排列相关的非均匀构型场),分别提出了它们的理论模型,继而对它们驱动的激子超快输运动力学过程进行了理论模拟.发现: 非均匀电场下,由于激子极化的正负电荷所处的电场强度不同,导致激子整体受到沿分子链方向的电场力,从而驱动激子超快输运; 非均匀构型场下,由于激子在不同耦合区域产生能的差异,诱导沿分子链方向激子输运的驱动力,从而驱动激子超快输运.本工作中,非均匀电场和构型场驱动的激子输运速度比由传统的Förster或Dexter机制主导的激子输运速度可分别提高1和2个数量级.这些结果丰富了大家对聚合物光伏体系内激子超快输运的认识,并从理论上提出了该体系内实现激子超快输运的新机制,为进一步提高器件的光伏效率提供了新的思路.
猜你喜欢
佛山科学技术学院学报(自然科学版)(2022年5期)2022-10-09
物理学报(2022年15期)2022-08-12
光子学报(2022年6期)2022-07-27
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
当代作家(2021年11期)2021-12-17
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
北京航空航天大学学报(2017年10期)2017-04-20
新高考·高一物理(2016年7期)2017-01-23
