
药品专利保护期补偿制度的中国路径
——《专利法修改草案(2019)》之完善
2019-10-23 06:31梁志文
法治现代化研究 2019年4期
梁志文
一、引 言
作为典型的TRIPs-Plus规则,专利保护期延长制度肇始于美国1984年《药品价格竞争与专利保护期恢复法案》(Hatch-Waxman法案)。该制度主要包括两种类型的保护期延长:专利授权审批过分延误的补偿与产品上市审批导致管制性延误的补偿。在美国法上,前者被称之为“专利保护期调整”(patent term adjustment,PTA),所有专利权人都可以主张补偿;后者被称之为“专利保护期延长”(patent term extension,PTE),专指药品等因上市审批时间而产生的专利保护期补偿或延长。(1)参见Lisa Clark, Debbie Beadle, Patent Term Extensions: Issues, Challenges and Implications for Pharmaceuticals, 1(4) Pharm. Pat. Analyst, 427, 432 (2012).专利保护期延长制度是美国近些年来通过双边或多边自由贸易协定试图推行的重要规则。(2)参见梁志文:《美国自由贸易协定中药品TRIPS-Plus保护》,载《比较法研究》2014年第1期。以最能体现美国意图的《美韩自由贸易协定》为例,其第18.8条第6款第1项规定了PTA,第2项即为PTE的规定:“对于受专利保护且在成员境内批准上市的新医药产品或新医药产品的制备或使用方法,应专利权人的申请,各成员将给予其专利保护期的调整,或对新医药产品及其制造、使用方法所涉及的专利权保护期予以延长,将其作为在成员境内首次上市许可审批导致专利有效期不合理缩短的补偿。本
节规定的所有调整应授予产品、制备或使用方法所涉专利的所有权项,也包括同样的权利限制与例外。”同时,该条注释还规定,“新医药产品”最少应包括在成员境内首次批准的包含新型化学实体(new chemical entity)的产品;“专利有效期”是指产品上市批准之日与原专利权保护期届满之日的期限。
2017年10月,中共中央办公厅、国务院办公厅发布《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(以下简称为《意见》),提出探索建立药品专利链接制度、保护期补偿制度和完善数据保护制度,在此基础上,国务院已经决定设置药品数据保护与专利保护期补偿制度。(3)参见《4月12日的国务院常务会定了这3件大事》,载“中国政府网”,http://www.gov.cn/xinwen/2018-04/13/content_5282188.htm,最后访问日期:2019年6月16日。专利法第四次修订草案,[以下简称为专利法(草案)],于2019年1月4日在“中国人大网”公布,并向广大社会公众征求意见,其第43条第2款规定了药品专利保护期补偿制度。该条规定:“为补偿创新药品上市审评审批时间,对在中国境内与境外同步申请上市的创新药品发明专利,国务院可以决定延长专利权期限,延长期限不超过五年,创新药上市后总有效专利权期限不超过十四年。”
然而,作为中国专利法的重大进展,药品专利保护期延长这一TRIPs-Plus规则的制定竟然在理论上未能得到充分的重视。(4)截至本文完稿之日,中国主流法学刊物(CSSCI法学类期刊)没有发表过专门以“专利保护期延长或补偿”为主题的论文,知识产权类和医药类的杂志发表了少量的研究成果。在专利法第四次修订时,专利无效程序、强制许可、当然许可以及赔偿制度都发表了较为深入的研究成果;但极少涉及保护期延长或补偿。这明显与药品专利的社会价值、经济价值并不相称。无论从理论重要性还是从产业价值来看,药品TRIPs-Plus规则的制定应该审慎,这是因为制药业的激励创新机制所具有的效果与其他产业迥异,它与公共健康密切相关。专利权人可以对专利产品采取垄断定价的策略,这是原研药产生巨额利润回报的原因。当然,垄断定价不只是制药业所采用,许多产业也采用该定价策略,如苹果公司iPhone X的定价。“但是,很少有产品像药品一样具有太多的正外部性,”“更重要的是,越是涉及生与死的事情,焦虑不安的人们就越有可能支付更高的市场价格。”(5)Amy C. Madl, Using Value-Agnostic Incentives to Promote Pharmaceutical Innovation 71 STAN. L. REV. 1305, 1309—1310 (2019).
本文以正在审议中的专利法(草案)为中心,以利益平衡为基本原则,运用比较法资料,研究药品专利保护期补偿制度的基本构成。笔者认为,现行专利法(草案)所规定的药品专利权补偿制度过于原则,其所规定的基本规则有重大缺陷,将会导致严重的利益失衡。因此,有必要进一步完善其具体规则。
二、药品专利保护期补偿制度中的利益分配
专利保护期为自申请日起的20年;但对于药品专利而言,权利人大都要花费10年左右的时间进行研发和临床试验,以获得药品监管部门的上市审批。原研药商认为,这实质性地缩短了药品专利权的有效期限,需要法律对其进行补偿。因此,药品专利保护期延长的最大理由是药品存在上市管制,相比不存在管制的其他产品,它使得药品专利的有效期要大大缩短。作为 Hatch-Waxman 法案的重要组成部分,原研药商常常将该制度与“Bolar例外”联系起来,认为专利法如果规定了“Bolar例外”,就必须要规定保护期延长制度,以实现法律之下的利益平衡。
然而,如果从整体来看药品创新保护制度对原研药研发的激励影响,原研药享有专利保护与数据保护的双重激励机制。Hatch-Waxman 法案还规定了保护药品上市测试数据或其他数据,在这一期限内,由于学名药商不能利用原研药商提供的数据来进行上市申请,实质性地延长了原研药的市场专有性。之所以保护原研药的数据,也是因为原研药获取药品的安全性、有效性和质量可靠性具有较高的研发成本和失败风险。(6)参见梁志文:《论TRIPS协议第39.3条之数据保护》,载《法治研究》2014年第2期。从这一层含义来说,药品专利权保护期的延长属于对上市管制所产生的成本之双重补贴。事实上,药品专利权保护期是否予以延长,并未严重影响到原研药商的整体利润。从整体来看,2016年世界500强中12家制药公司的平均净利润率(23.58%)是其他所有企业(5.46%)的4倍多,其平均净资产收益率(11.2%)与其他所有企业(3.03%)的差别也高达3倍多。(7)参见梁志文:《论以信息公示为中心的药品专利链接制度》,载《中国专利与商标》2018年第1期。
美国学者的研究表明,在通过1984年Hatch-Waxman 法案之前,美国制药业的利润并未有降低的现象,反而一直处于不断提高之中。美国1981年发布的一份报告就指出:“自20世纪50年代以来(即实施药品上市管制以来,笔者注),在美国所有主要产业中,制药业属于最有利可图的产业部门。股东投资于该产业的税后利润回报率稳定在一个较高的水平,并且超出所有产业的税后投资回报率。”(8)Albert Gore,Jr., Patent Term Extension: An Expensive and Unnecessary Giveaway, 2 HEALTH AFFAIRS 25, 26 (1982).同样,该时期制药业的研发投入也并未呈现出降低的态势,如《财富》杂志1981年10月19日报道:“默克公司该年在药品研发方面投入了28亿美元,是10年前的4倍。礼来公司在1980年投入的研发资金是21亿美元,比1971年超出3倍还多。辉瑞公司的投入大约在18亿美元左右,1970至1980年间的年增长率高达16%。施贵宝公司最近5年的投资也增长84%,达到9.1亿美元。”(9)see note⑧,P27。在这一背景下,如果广大发展中国家移植美国Hatch-Waxman 法案,制定药品专利权保护期延长的制度,对原研药商而言绝对是一个重大利好,也是其获得额外利润的重要保障。
很明显,将药品专利保护期予以延长将减轻原研药的竞争压力,有利于提高其利润回报。但另一方面,它将延缓仿制药的上市,不利于药品价格的控制,也将影响患者对药价的可承受性,进而影响药品可及性。为了在药品专有权保护期内获得最大的利润,原研药商通常在知识产权保护制度下使用撇脂定价策略,以获取药品的最大利润。所谓撇脂定价,是指将药品以极高的价格销售给患者,如诺华公司的“格列卫”在中国的定价曾经高达每盒23500元,是印度仿制药每盒200元的117倍。撇脂定价策略之所以能在原研药领域得以普遍实施,其原因不仅在于原研药的开发成本高、价格弹性小,还在于患者对救命药的需求强烈、对价格不敏感,更在于市场上缺乏足够具有竞争力的同样药品。“格列卫”就是这样的救命药,它居然把绝症——慢性粒细胞性白血病(血癌的一种)变成了吃药就能控制的慢性病,从而挽救了成千上万的病患,它成为病患生存的必需品。此时,药品售价可依患者最大支付能力为标准来制定。“格列卫”不只是在中国售价昂贵,在美国也同样采取撇脂定价策略。尽管这一定价策略导致许多患者付出了巨额的医疗费用,甚至因病堕入穷困而被指责“太不人道”,但它为诺华公司带来了丰厚的利润回报,为其后续研发积累了巨额的资金。(10)叶盛:《为什么格列卫会这么神?这么贵?》,载“凤凰网”, http://news.ifeng.com/a/20180709/59068670_0.shtml,最后访问日期:2019年6月7日。
“中国在完善居民医疗保障体系方面成绩斐然,但无须讳言,其仍处于较低水平的状态,药价往往成为影响药品可及性的重要因素。对于受专利保护的原研药来说,在发达国家和发展中国家的售价可能并无实质性区别,即使发展中国家的患者只有少数人购买得起高价药。”(11)参见前引⑦,梁志文。因此,中国专利法如何移植药品专利保护期补偿制度,这一问题值得审慎考虑。确如有学者所指出的,“药品延长专利保护(期)是一把双刃剑,掌握推出该制度的时机非常重要。”(12)何炼红、鲁浪浪:《中国医药发明专利试验例外制度研究》,载《时代法学》2009年第6期。该时机的出现,可能取决于两个因素。一是中国在原研药产业有了实质性的发展之后,需要在国际去开拓市场,需要在法律上确保创新成果的投资回报。二是在面临美、欧的贸易谈判压力时,作为一种不同产业折中的安排,也可以引入该制度。在后一情形下,则应该在谈判的协议中为国内立法争取充分的弹性空间,以维系公共健康、药品可及性与产业发展之间的平衡。
三、药品专利保护期补偿的实体条件
药品专利保护期补偿制度主要由三部分内容组成:补偿条件、期限计算与基本程序。很明显,专利法(草案)第43条第2款的规定过于原则,仅规定了补偿条件与期限计算。其中,该条规定的药品专利保护期补偿的实体条件是:“为补偿创新药品上市审评审批时间,在中国境内与境外同步申请上市的创新药品发明专利。”但是,该规定的有些内容既不科学,也不合理。
(一)需要修订的实体条件
根据专利法(草案)第43条第2款规定,药品专利保护期补偿的实体条件包括四方面:
第一,可获得专利保护期延长的客体只能是“药品”发明专利,不包括用途专利和制备方法专利,也不包括医疗设备的发明专利,同样不包括实用新型专利。从比较法来看,该规定所保护范围确实相对较窄。例如,美国专利法第156条第a款规定,“产品、使用产品的方法或制造产品的方法之专利”,在符合其他条件时可以申请延长其保护期。日本专利法第67条第2款规定,“专利发明的实施”需要取得行政许可的话,在符合其他条件的情况下,可以延长其保护期。中国台湾地区所谓“专利法”第53条第1款规定,可以申请延长的专利权包括“医药品、农药品或其制造方法发明专利权”,但第3款规定“不及于动物用药品”。但是,笔者赞同仅对产品发明专利予以延长、不延及方法专利,其理由在于我国在医药产业还不宜实施过高的保护标准,以免影响公众的药品可及性。
第二,它仅涉及“创新药品”的发明专利权保护期延长。专利法(草案)并未明确“创新药品”的概念。但根据《药品注册管理办法(修订征求意见稿)》(2017年)第12条规定,“化学药品注册分类可分:创新药,改良型新药,仿制药。”“生物制品注册分类可分为:新型生物制品,改良型生物制品,生物类似药。”同时,《药品试验数据保护实施办法(暂行)(征求意见稿)》(2018年)第3条中也使用了“创新药、创新治疗用生物制品”的概念。因此,它应该不包括新型生物制品。即,创新药品是指包含有新型化学实体的药品。从比较法来看,它的保护范围也非常狭窄。例如,欧盟《专利补充保护证书(Supplementary Protection Certificate,SPC)条例(第469/2009号条例)》(以下简称《条例》)第1条对“药品”或“产品”的定义也并未仅限定为化学药。澳大利亚专利法第70条规定,“医药物质”专利权人在符合条件下,可以申请专利权保护期的延长。
药品专利保护期补偿制度应该延及新型生物制品。首先,尽管现阶段生物制品在药品中的比重并不大,但在处方药市场上,它日益成为最为重要的组成部分。生物制品大都是来源于活体材料的药品,包括来源于血液的产品、疫苗和大多数蛋白质产品。与传统化学药相比,生物制品的全球市场销售额持续快速递增。在2000年,生物制品的销售量占全球销售量最大的100种药品中的11%;而到2014年,在销量最大的10种药品中,将有7种为生物制品,销量最大的100种药品中,生物制品占到50%;(13)参见Sarah Sorscher, A Longer Monopoly for Biologics: Considering the Implications of Data Exclusivity as a Tool for Innovation Policy, 23 HARV.J.L &TECH. 285, 285-286 (2009).2018年最畅销与销售额增长最大的十大药品全都是生物制品,预计2022年全球的生物药市场将高达3260亿美元。(14)参见《2022年全球生物药市场达3260亿美元,预计2018年单抗类药物崛起成霸主》,载“中国生物技术信息网”,www.biotech.org.cn/information/153704,最后访问日期:2019年6月7日。其次,生物制品的研发成本更高。通常认为,生物制品的开发不同于小分子化学药品,它更为复杂。DiMasi(2007)估算的生物制品研发成本达到13.18亿美元,远远高于化学药。(15)参见J. A. DiMAS, H. G. Grabowski, The Cost of Biopharmaceutical R&D, 28 MAMANGE DECIS. ECON. 469 (2007).第三,生物医药产业属于国家重点发展的产业。2017年1月,国家发改委正式印发《“十三五”生物产业发展规划》,生物医药产业是其重要组成部分,该规划特别强调要重点发展生物技术药物、新疫苗、新型细胞治疗制剂等多个创新药物品类。
因此,应该删除“创新”一词;“药品”的概念涵盖了化学药和生物制品,还能够延及中药等领域的发明专利权,这也符合中国发展中医药产业的基本政策。《中华人民共和国药品管理法》第102条规定:“药品,是指用于预防、治疗、诊断人的疾病,有目的地调节人的生理机能并规定有适应症或者功能主治、用法和用量的物质,包括中药材、中药饮片、中成药、化学原料药及其制剂、抗生素、生化药品、放射性药品、血清、疫苗、血液制品和诊断药品等。”当然,专利法(草案)中的“创新”一词也有可能体现的是法律起草人将其保护范围限定于新药的意图,或者并未理解“创新药”在药品管理法中的特定含义。如果这确实是起草人对“创新”一词所用的含义,则该词明显属于赘语,应予删除。因为发明专利必然具有新颖性,而申请专利药的首次上市也必然是新药的上市。这也是美国、日本和中国台湾地区等所谓“专利法”上并未规定“新药”或“创新药”这一条件的原因。
第三,权利人必须“在中国境内与境外同步申请上市”。这一规定具有中国国情,其重要考虑是将其视为创新驱动发展战略下国家鼓励医药领域的投资并提升产业技术发展的方法。一方面,TRIPs-Plus规则被视为鼓励制药技术开发、激励跨国制药公司尽快在中国上市新药的“胡萝卜”。因为“近10年来,中国上市的一些典型的新药时间平均要比欧美晚5年至7年,国外都已经用了六七年了,中国才上市,这是因为很多制度设计造成了新药在中国上市慢半拍。”(16)《让百姓用得起更多救命药放心药》,载“中国政府网”,http://www.gov.cn/zhengce/2017-10/10/content_5230469.htm,最后访问日期:2019年6月7日。另一方面,TRIPs-Plus保护规则还被视为惩罚原研药延迟进入中国市场的“大棒”,其主要手段是设定限制知识产权保护的具体条件。例如,类似于专利实施义务,获得专利保护期的延长需要品牌药商承担在中国的实施义务或供应义务。但是,这一规定具有很大局限性——不利于首先在中国上市的原研药厂家。它既不利于跨国制药企业在中国首先推出原研药,而且也将迫使中国的原研药厂家在多国同步上市其开发的原研药,这无疑会增大中国原研药厂家受保护的成本。而且,从某种程度上讲,此种情况下所限制的对象将会主要是中国的原研药厂家。因此,该条应该修改为“对在中国境内首先申请上市或在中国境内与境外同步申请上市的药品发明专利”。
第四,它系为了“补偿创新药品上市审评审批时间”。因此,能够获得专利保护期延长的药品专利必须经过药品监管部门的上市审批,且已获得上市批文。然而,专利法(草案)的表述也同样存在一些问题。从字面意义来看,它强调了药品专利保护期补偿制度的立法目的。这其实也是多余的,甚至有歧视其他受管制的产品、有违TRIPs协议第27条第1款不得歧视技术领域之嫌。例如,农药的上市也受管制,也会产生缩短其专利有效期的法律效果。中国台湾地区所谓“专利法”第53条就将保护期补偿的对象延伸到了农药。美国专利法第156条也并未宣示它仅是对药品上市评审审批时间的补偿,该条第g款6项c目规定了如果属于新兽药或动物用生物制品的话,其补偿期限不得超出3年。
大部分国家或地区的法律要求申请专利保护期补偿的药品通过了上市主管部门的审批。质言之,如果未经上市审批的药品专利权人,无权要求延长其保护期。例如,美国专利法第156条第a款第4、5项明确规定:“该产品在上市或使用之前须经过一定管制审查期限。”“除本项另有规定之外,产品上市或使用经过管制审查期限而被批准,且须为依法核准的首次许可。”欧盟《条例》第2条也明确规定:“在成员国境内受专利保护的产品,作为药品上市前须依欧盟委员会第65/65/EEC4号指令或第81/851/EEC5指令申请并获得行政许可,如符合本条例的条件,属于证书保护的对象。”日本专利法第67条之2第3款规定,“专利权期限延长注册的申请书,应在取得第67条第2款规定的许可后”方可递交。中国台湾地区所谓“专利法”第53条也有类似规定:“依其他法律规定,应取得许可证者,其于专利案公告后取得时,专利权人得以第一次许可证申请延长专利权期间。”
(二)必须补充的实体条件
专利法(草案)并未规定批准上市的药品与药品专利权之间的关系。而大多数国家的专利法都有类似规定,因为这一规定具有重要意义。改良型新药和改良型生物制品也按新药申请,需要履行上市评审审批手续,它是对已知活性成分的再创新,如在结构、剂型、处方工艺、给药途径、适应症等方面进行的改进,且具有明显临床优势。改良型药品本身可能不受专利保护,但它落入了基础专利(活性成分)的保护范围。此时,改良型药品的上市许可是否可以主张基础专利的保护期延长?或者说,基础专利的保护期可否多次得以延长?
首先,许多国家将包含受专利保护物质的药品之首次上市作为保护期补偿的条件。澳大利亚专利法第70条强调,“(获得上市批文的)医药物质本身须在专利说明书中所披露的物质中,且须落入某一权利要求或说明书所支持的权利要求之保护范围”;还须属于“该物质首次被批准”上市的情形。欧盟也有类似规定,其《条例》第3条d款规定:“‘作为药品上市获得批准’,是指该产品的首次批准。”第1条的定义中,“药品”是指“物质或物质组合”,“产品”是指“活性成分或活性成分组合”。美国专利法第156第a款第5项强调了“依法核准的首次许可”。因此,专利法(草案)应该规定“首次许可”条件,将改良型药品的上市审批情形排除出专利权保护期延长之外,从而避免药品专利权人采取常青树策略,通过改良型药品的上市来不断延长基础专利的保护期。
如何判断批准上市的药品是否属于受专利保护的物质?这是最易发生争议的、需要解释的法律条款。欧盟法院(Court of Justice of the European Union,CJEU)于1999年就在Farmitalia案(C-392/97)中对“产品受有效的基础专利保护”这一条件作出了裁定,(17)参见Judgment of the Court (Fifth Chamber) of 16 September 1999—CASE C-392/97,http://curia.europa.eu/juris/liste.jsf?language=en&jur=C,T,F&num=C-392/97&td=ALL.,最后访问日期:2019年6月27日。涉案的基础专利为一项德国专利,其权利要求保护的是去甲氧基柔红霉素,而不包括其盐,但对包括盐酸伊达比星在内的去甲氧基柔红霉素和盐都申请了SPC。CJEU认为欧盟内部没有统一的专利法,产品是否受基础专利保护这一问题属于成员国国内法的范畴。此后,不同法院就此发展出两项不同的判断方法:(1)侵权测试标准,即SPC申请中的产品之制造、销售是否侵犯基础专利权;(2)披露测试标准,即SPC申请中的产品是否在基础专利的权利要求字义范围之内。判断标准存在差异这一现实并不符合欧盟法律协同的初衷。2011年CJEU试图在Medeva案(C-322/10)中作出调整。CJEU否定了“侵权测试”标准,认为受基础专利保护的“活性成分”必须在“权利要求的字义中明确特定化”才能申请SPC。(18)参见Franz-Josef Zimmer et.al., Recent Decisions of the European Court of Justice of the European Union on Supplementary Protection Certificates: A Few Answers—Many Questions, 33 Biotechnology Law Report 172 (2014).
在日本法上,对同一活性成分、适用症或疗效的不同给药形式、剂量等,不能享有额外的延长期限。但是,属于同一活性成分但不同适用症或疗效的药品,可以批准其延长。在权利要求范围之内的不同活性成分,但不包括不同的盐,可以要求获得额外的延长。2015年11月,日本最高法院就基因科技公司(Genentech Inc.)贝伐珠单(阿瓦斯汀)抗癌药的专利保护期延长申请被特许厅驳回一案作出了终审裁决。(19)参见Takeshi S Komatani, Patent Term Extension: The Supreme Court of Japan Gave An Answer to The Question and The JPO Now Has to Prepare New Examination Guidelines, 5(3) Pharm. Pat. Anal. 155—156 (2016).在该案中,日本特许厅依据2011年修订的《专利审查指南》驳回了基因科技公司的申请案,该指南规定,如果早先获批的药品与申请延长的获批药品从专利权的范围来看并无区别,则将驳回专利权期限延长的申请。质言之,即使两者之间在剂量、给药途径等方面存在不同,但如果专利权利要求与药品的活性物质等差异无涉,则将驳回申请。日本最高法院认为,应以专利权保护期延长申请案的客体相关的处置(如药品上市许可)与在先的处置(批准)相比较的结果,判断药品是否实质一致,就该申请案之专利发明的类型等来看,如果在先批准的药品包含了申请案中药品的制造和销售行为,正确的解释是:对申请案客体的处置并不是实施申请案专利所必需的审批。但是,该案在先获得的批文并没有影响在后批文以新药形式获批的事实表明,后者仍然有权获得保护期之延长。2016年4月,日本特许厅依据该案修订了《专利审查指南》。(20)参见Japan Patent Office (Japanese). http://www.jpo.go.jp/shiryou/kijun/kijun2/h2803_kaitei.htm,最后访问日期:2019年6月7日。
笔者赞同药品专利保护期补偿制度的保护对象仅限于药品、不及于方法(新用途)或制备方法专利。因此,笔者认为,中国不应移植日本的做法,应坚持同一活性成分只能获得一次保护期的延长。
其次,同一专利权的保护期只能延长一次。美国专利法第156条a款2项规定保护期延长的条件之一是“专利权期限并未依本节第(e)(1)条规定而延长过。”欧盟《条例》第3条第c款也有类似规定,并引发了广泛争议的法律问题。CJEU在1996年裁决的Biogen案(C-181/95)中认为,该款不得解释为“不允许对同一产品授予多份SPC”,只要该产品受多项由不同权利人持有的基础专利保护;该款的含义是,仅一项证书可(may)授予给每一项基础专利(each basic patent)。因此,这被欧盟成员国的专利局解释为:受同一项基础专利保护的多个产品可以获得多份SPC。但是,CJEU在Medeva案(C-322/10)中发生了重大的改变,认为即使不同的产品获得了上市许可,但每项专利不得授予多份SPC。尽管有观点认为Medeva案的基本事实与Biogen案不同,但大多数成员国的专利局都开始实施“一项专利一份SPC”(one SPC per patent)的要求。当然,也有部分专利局坚持“每项专利每项产品一份SPC”(one SPC per product per patent)的原有规则。(21)参见Franz-Josef Zimmer et.al., Recent Decisions of the European Court of Justice of the European Union on Supplementary Protection Certificates: A Few Answers—Many Questions, 33 Biotechnology Law Report 173 (2014).
第三,该药品所涉及的基础专利仍然处于有效保护期之内。质言之,如果批准上市的药品已经保护期届满,或者被宣告无效,则不能够申请保护期延长。美国专利法第156条a款第1项明确规定,递交药品专利“保护期延长的申请之前,专利权期限并未届满”。欧盟《条例》第3条第a款也明确了“产品受有效的基础专利保护”。
第四,保护期获得延长后的专利权之内容。欧盟《条例》第5条规定:“SPC享有与基础专利一样的权利,也与基础专利一样受到限制、承担义务。”这也大体上属于各国法律的共同规定。但是,SPC与专利权的保护范围有着重要不同。《条例》第4款规定:“在基础专利所保护的范围之内,证书的保护仅延及作为药品上市的批文所指的产品,以及在证书期限届满之前对作为药品的产品之所有利用行为。”在本质上,SPC并非是专利权本身的保护期延长,而是一种经申请而被批准的市场独占权,即,给予通过批准的药品及其使用的一段时期的市场独占权。在SPC有效期内,可以进行专利药的仿制,但学名药不能上市销售和使用。
四、药品专利保护期补偿的期限计算
专利法(草案)规定药品专利“延长期限不超过五年,创新药上市后总有效专利权期限不超过十四年”。这一规定与绝大多数国家的法律基本一致,但过于原则而导致可操作性仍然不够。其中,它并未界定何谓“总有效专利权期限”,也未界定不超过5年的计算标准。从比较法的角度来看,各国规定的期限计算方法虽略有不同,但大都明确规定了可操作性的具体规则。
(一)美国
在美国,药品专利期延长的具体期限取决于法律规定的三个期间。一是药品注册时美国联邦食品药品监督管理局(FDA)在强制审查程序中所耗费的时间或期间,即管制性审查期间(Regulatory Review Period,RRP)。一般来说,RRP包括试验阶段和审批阶段两部分。人用药品的试验阶段是从研究用新药申请(INDA)被批准,到新药申请(NDA)初次递交之间的期间。审批阶段是从NDA递交到获得批准之间的期间。二是法律明确规定的两项限制:(1)不得超过5年,或法律特别规定的2年(第156条g款6项C目规定的法律生效前申请但尚未在生效日获批),或3年(即第156条g款6项C目规定的新兽药或动物用生物制品)。(2)药品注册申请被批准后剩余的专利权期限(即专利有效期限)与延长期限之和不得超出14年。三是规定了申请人的合理勤勉义务。美国专利法第156条c款1项规定:“专利权期限延长的申请人在管制审查期间内并未履行合理勤勉(due diligence)义务而产生的期间,依本条第(d)(2)(B)节规定,应予以扣除。”
据此,美国法上专利保护期补偿的具体计算,分两步来确定。第一步,确定“可以合格延长的专利期限”(Patent Term Eligible for Restoration,PTER),其计算公式如下:(22)参见Jaime F. Cardenas-Namat, Thirty Years of Flawed Incentives: An Empirical and Economic Analysis of Hatch-Waxman Patent-Term Restoration, 29 Berkeley Tech. L.J. 1301, 1318 (2014)
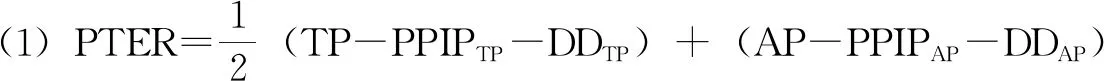
时间单位:日
该公式中,TP是指“测试阶段”(Testing Phase),PPIP是指“专利授权前的阶段”(Pre-Patent Issuance Phase),DD是指“申请人未能履行合理勤勉义务的所有期间”(Due Diligence),AP是指“许可审批阶段”(Approval Phase)。下标的TP、AP分别表示的是“测试阶段”和“许可审批阶段”产生的各种期间。
第二步,计算 “最终得以延长的期限”( Patent Term Restored,PTR)。它分两种情况:第一,如果PTER小于或等于5年(或第156条g款6项C目规定的2年或3年);同时,PTER与药品注册申请被批准后剩余的专利权期限(Effective Patent Term,即有效专利期)之和小于或等于14年,则最终得以延长的期限就是PTER。如果大于14年,则最终得以延长的期限是14年减去有效专利期的值。第二,如果PTER大于5年(或第156条g款6项C目规定的2年或3年);则PTER仅限于5年(或第156条g款6项C目规定的2年或3年),再依据第一种情况确定最终得以延长的期限。
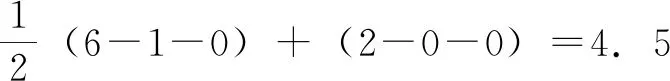
(二)欧盟
与美国法的计算方法不同,欧盟补充保护证书(SPC)的有效期限为专利药品因上市受到的减损期间,但其期限自基础专利期届满之日起不得超过5年,且药品通过批准后剩余的基础专利保护期加上SPC的有效期不得超过15年。具体计算方法也分两步。第一步确定“可以延长的保护期限”,其标准是:“基础专利申请日至共同体内产品首次上市被批准之日(PA),再减去5年期限”;且“不得超出自其生效之日起的5年”。即,超出者不得计入。第二步确定“最终授予的SPC期限”,其标准是:根据第一步确定的“可以延长的保护期限”,加上“药品批准后剩余的基础专利保护期”(即“有效专利”),如果其总和不超过15年,则“可以延长的保护期限”即为“最终授予的SPC期限”。如果其总和超出15年,则超出的期间不计入最终授予SPC期限。
(三)日本
日本《专利法》第67条第2款规定可延长的期限最长为5年,但在具体计算方式上,它不同于美、欧规定的专利权有效期与被延长期限的总和不得超过14或15年,日本专利法并未有该限制,故其基础专利的保护期与被延长的期限之和可能会超出15年。如下图所示,合格的可被延长的期间等于因产品上市管制而不能实施发明专利的期间。该期间始于临床试验被批准或专利登记之日,两者以后至者为准;终止于产品上市批文被发往申请人处之日。递交研究用新药申请(IND)之日视为临床试验被批准之日;专利登记日是指专利授权且向特许厅缴纳了注册费之日。专利注册之日常常为专利授权日,专利公报发行日并不影响该期限的计算。上市批文发往申请人处之日,则常常以申请人实际收到批文之日为准。(23)本节内容参考日本专利审查指南之英译本而撰写,英译本指出,如有歧义,应以日文版为准。https://www.jpo.go.jp/e/system/laws/rule/guideline/patent/tukujitu_kijun/document/index/09_0100_e.pdf,最后访问日期:2019年6月7日。
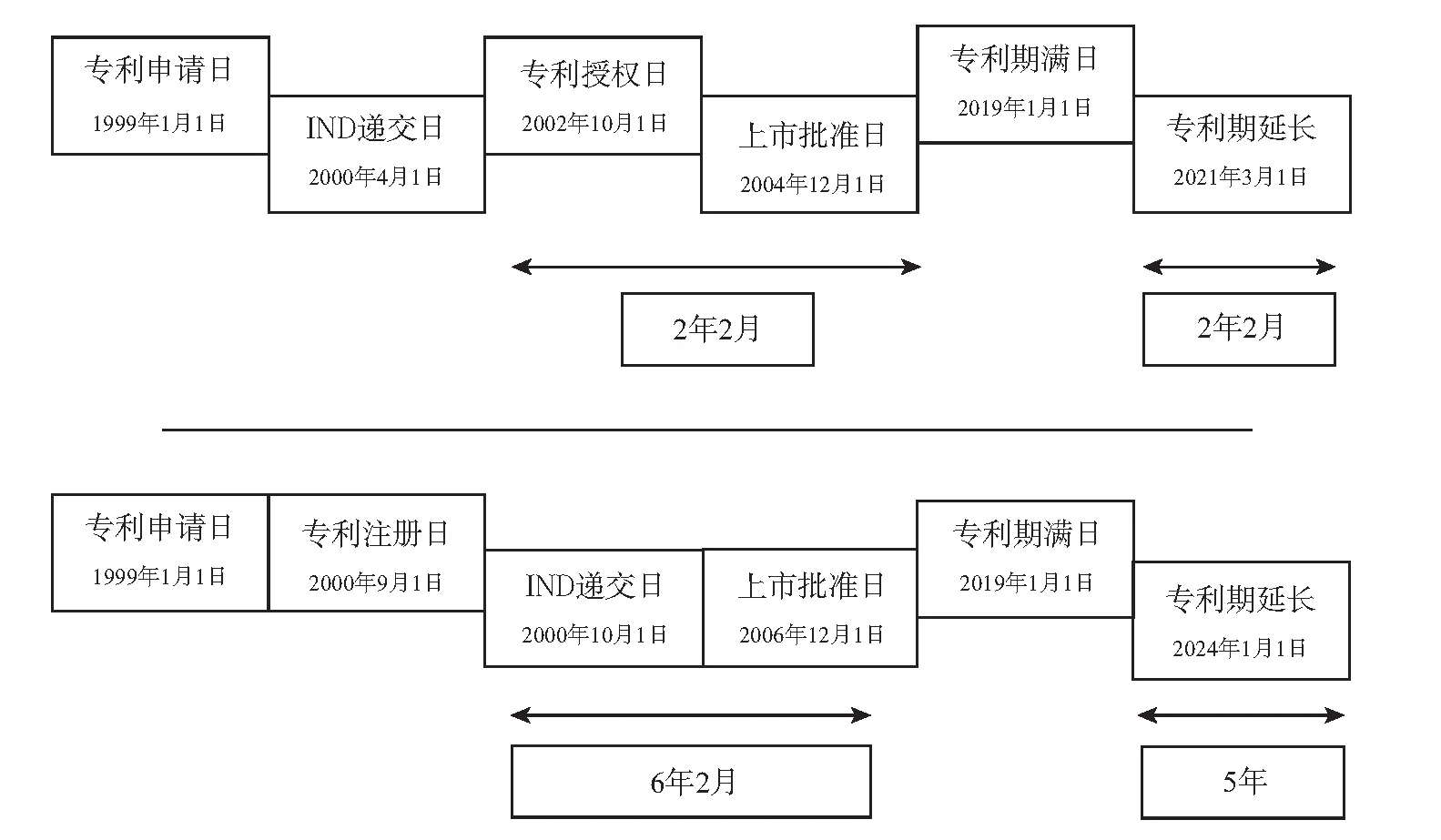
日本专利法上的期限延长
(四) 专利法(草案)的期限计算
专利法(草案)仅参照美国法规定了期限计算的基本原则,但并未明确其具体计算的方法。笔者认为,从计算简便及立法目的考虑,中国专利法在计算方法上应借鉴日本法的规定,即合格的可被延长的药品专利期等于因产品上市管制而不能实施发明专利的期间。其次,为了激励原研药商及早在中国申请上市,明确专利权人在申请药品上市方面应负有合理勤勉义务具有重要意义。所谓“合理勤勉义务”,是指“在管制审查过程中依通常人之合理预期并通常采取的行为、注意程度、持续的直接努力和适时性。”(24)美国专利法第156条d款第3项。因此,被延长的专利期应减去权利人未有正当理由而延迟申请上市的期间。
五、药品专利保护期补偿的程序规定
专利法(草案)缺乏明确的程序规定。药品专利保护期补偿涉及药品上市审批的行政主管部门与专利审查的行政主管部门之间的协作。因此,相关程序的规定非常重要,它是该制度能够得以顺利运作的关键之一。专利法(草案)将保护期补偿的裁定权规定为“国务院”(“国务院可以决定延长专利权期限”),这会使得该制度的实施面临一定困难,因为药品专利保护期补偿制度一旦确立,根据前述规定的补偿条件,肯定不会是像药品专利强制实施许可那样只存在个案申请现象。如此一来,国务院不可能对大量的药品专利保护期补偿申请案作出受理审查的决定,而只能由专利审查部门或药品监管部门来作出裁定。
专利审查部门作出延长药品专利期的决定必须依据药品上市审批部门的审查决定,故必须在法律上建立两者之间的沟通渠道。例如,我国台湾地区所谓“专利法”第53条第5款规定:“主管机关就延长期间之核定,应考虑对国民健康之影响,并会同中央目的事业主管机关订定核定办法。”在美国法上,美国联邦专利商标局(PTO)和FDA是分工合作的关系。从分工来看,关于专利权期限延长是否符合条件,由FDA作出决定;而关于专利权效力和保护范围的法律问题,由PTO作出决定。同时,两者之间又是紧密合作的。申请人在收到FDA批准函60天内向PTO递交专利期延长申请,PTO在收到申请的60天内将申请送交至FDA,由后者负责计算管制审查期,并将计算结果通知PTO。虽然机构改革后,国家知识产权局与国家药品监督管理局均隶属于国家市场监督管理总局,但也同样存在内部协调的问题需要法律明确规定。
药品专利保护期补偿制度应明确规定申请人资格与申请时效。从比较法来看,各国法律大都规定具有申请人资格的只能是专利权人。如美国专利法第156条a款3项规定:“在符合本节第(d)条1至4段的条件下,由专利权人或其代理人递交期限延长的申请。”同时,该条还规定,专利权人必须在药品获得FDA批准上市后的60天内向PTO递交专利期延长之申请。欧盟《条例》第6条规定:“证书将授予基础专利的持有人或其继承人。”第7条规定:“证书的申请须在获得第3(b)条之药品上市批文之日起6个月内提出。”“但如果上市批文在基础专利授权日前即已获得,证书的申请必须在专利授权之日起6个月内提出。”在日本法上,申请人须以专利权人的名义递交;如果属于共有的专利权人,则必须共同提出申请,否则将会被驳回。如果上市批文的获得者并非是专利权人,则其必须属于在特许厅登记的被许可人;至于获得的许可权性质,它既可以是独占许可,也可以是普通许可。此外,许可合同的登记可以在专利保护期延长申请之后递交,但必须在特许厅批准专利保护期延长申请案之前完成。在获得上市批文后,专利权人必须在收到批文之日起的3个月内且在专利权期满之前向特许厅递交申请;如果在专利权期满前6个月难以得到批文的,则应事前向特许厅提出临时申请以告知审批事实,但临时申请并不延长前述规定的期间。
药品专利保护期补偿制度应明确规定第三人参与程序。在美国法上,药品保护期补偿的具体期限需要扣减权利人未尽合理的勤勉义务时所延误的时间,为此,它规定了具体的专利权期限延长的异议程序。在FDA公布管制审查期决定后的180天之内,任何人可以向FDA提出尽责申诉(Due Diligence Petition),目的是确认申请人未能尽责,由FDA作出减少专利期延长期限的决定。同时,在FDA公布尽责决定后的60天内,任何人都可以要求FDA举行尽责听证(Due Diligence Hearing),以确认申请人是否履行合理的勤勉义务。
药品专利保护期补偿制度还应明确规定审理程序与无效程序。一般来说,它基本与专利程序相同。例如,欧盟《条例》规定,SPC的批准与管理隶属于批准其基础专利的成员国专利局或成员国法律授权的有关部门,申请人须向各成员国专利局递交申请文件,获得的SPC也仅在该成员国有效。对于被驳回的SPC申请,成员国应当提供与专利申请审查和上诉程序相同的国内法程序。
六、结 论
在中国知识产权立法实践中,专利法等法律文本一般具有原则性规范的特点,其具体落实措施常常由《实施条例》或《细则》来完成。但是,如果某些规则涉及一项制度的基本构成要件,则不宜在立法层级较低的《实施条例》或《细则》等行政法规中规定。药品专利保护期补偿制度可在《专利法》上作出原则性规定,其具体落实由《专利法实施细则》与《专利审查指南》来实现。但是,构成保护期补偿制度的基本构成要件的法律规则同样不宜在《专利法实施细则》或《专利审查指南》中规定。专利法(草案)第43条第2款的规定存在两方面的缺陷:第一,缺乏审查程序、没有规定批准上市的药品与药品专利权之间的关系以及保护期延长法律效力,因此应该予以增补;第二,部分法律规则的表述不科学,不利于促进中国原研药的自主开发,应该予以修订。
具体而言,笔者认为,专利法(草案)第43条第2款应予以独立,并作如下修订:
对在中国境内首先申请上市或在中国境内与境外同步申请上市并获得批准的药品发明专利权人,可在专利有效期内、以首次获得的许可证向国务院专利行政部门申请延长专利权保护期限,但该申请仅以一次为限。核准延长的期限为国家药品监督管理部门审批该药品上市申请的期间,但不得超出五年,且应扣除专利权人在药品上市审查期间内未履行合理勤勉义务而产生的期间;同时,药品注册申请被批准后剩余的专利权期限与延长期限之和不得超出十四年。
保护期依本款得以延长的专利权,在被延长的期限内其权利仅限于对产品被批准的许可范围内的利用行为。
申请药品发明专利保护期的延长,应递交申请书,附具证明文件,于首次取得许可证后三个月内,向国务院专利行政部门提出。国务院专利行政部门会同国家药品监督管理部门制定具体核准办法,符合条件的,由国务院专利行政部门予以公告。
猜你喜欢
英语世界(2022年9期)2022-10-18
中国医院用药评价与分析(2022年5期)2022-06-23
家庭医药·快乐养生(2022年6期)2022-06-23
大众健康(2019年6期)2019-06-10
世界知识(2017年21期)2017-12-28
知识产权(2017年11期)2017-01-26
知识产权(2016年6期)2016-12-01
知识产权(2016年5期)2016-12-01
知识产权(2016年3期)2016-12-01
