
奥拉西坦胶囊中主要未知杂质的鉴定及其含量测定
2020-04-08 01:21张菁孙婷赵昌梦姜建国
中国药房 2020年6期
张菁 孙婷 赵昌梦 姜建国

ABSTRACT OBJECTIVE: To identify the main unknow impurity of Oxiracetam capsule and determine its content, so as to improve the standard of quality control. METHODS: Two-dimensional UPLC-IT-TOF-MS was adopted to qualitatively analyze the unknown impurity. One-dimensional liquid chromatogram analysis was performed on ST PAK C18 ES column with mobile phase consisted of 0.02 mol/L sodium dihydrogen phosphate solution at the flow rate of 0.5 mL/min. The column temperature was set at 30 ℃, sample size was 20 μL. The detection wavelength was set at 210 nm. Two-dimensional liquid chromatogram analysis was performed on Techmate C18-STⅡ column with mobile phase consisted of 0.02 mol/L ammonium acetate solution at the flow rate of 0.5 mL/min. The column temperature was 30 ℃. Mass spectrometry was adopted (electropray ionization source, MS+ and MS- mode data acquisition). After the target impurity was located by one-dimensional liquid chromatography, it was transferred to two-dimensional liquid chromatography-mass spectrometry system for qualitative analysis. The unknown impurity structure was inferred by means of molecular formula prediction module “Accurate Mass Calculator”in LCMS Solution, and the refined impurity products by preparation and purification were standardized and confirmed. The impurity content was determined by HPLC (with the same condition of one-dimensional liquid chromatography for qualitative analysis). RESULTS: The main unknown impurity in Oxiracetam capsules is oxiracetam acid. The content of the refined product was 99.5% after preparation and purification. The contents of oxiracetam acid in 9 batches of Oxiracetam capsules were 0.05%-0.14%. CONCLUSIONS: The established two-dementional UPLC-IT-TOF-MS method can accurately locate the peak position of the impurity oxiracetam acid, and analyze its structure, while the corresponding content determination method can better separate the impurity from the main drug and other components, with good sensitivity, precision, repeatability, stability and accuracy. The quality of the finished product of Oxiracetam capsules can be well controlled by using above method.
KEYWORDS Two-dementional UPLC-IT-TOF-MS; HPLC;Oxiracetam capsule;Oxiracetam acid; Impurity; Identification; Content determination
奧拉西坦为吡拉西坦的类似物,其化学名为4-羟基-2-氧代-1-吡咯烷乙酰胺(化学结构式见图1),临床上主要用于改善老年性痴呆和记忆障碍患者的记忆功能[1],其作用机制是通过促进磷酰胆碱和磷酰乙醇胺的合成,提高大脑中腺苷三磷酸(ATP)/腺苷二磷酸(ADP)的比值,增加大脑中蛋白质和核酸的合成[2]。按照2015年版《中国药典》(四部)相关指导原则[3]开展的稳定性试验结果显示,奥拉西坦胶囊在贮存过程中杂质含量有逐渐增加的趋势。奥拉西坦原料药现行注册质量标准对其有关物质采用高效液相色谱(HPLC)自身对照法进行测定[4-5],但无法对有关物质的含量进行准确控制;而奥拉西坦胶囊质量标准则未对有关物质进行控制[6]。基于此,本课题组参考相关文献[7-8],建立了超高效二维液相色谱-离子阱-飞行时间质谱法,对奥拉西坦胶囊中的主要未知杂质进行定性分析,确定杂质的结构,并对该杂质进行含量测定,为提高奥拉西坦胶囊质量控制标准、确保其临床使用的有效性和安全性提供技术支持。
1 材料
1.1 仪器
2D-LC-IT-TOF/MS型超高效二维液相色谱-离子阱-飞行时间质谱系统、SPD-M20A型二极管阵列检测器(用于定性分析)、LC-20AD型液相色谱仪(用于含量测定)均购自日本Shimadzu公司;Mercury-400型核磁共振(NMR)仪(美国Varian公司);CP225D型十万分之一电子天平(德国Sartorius公司)。
1.2 药品与试剂
奥拉西坦胶囊(厂家A,批号:20180701、20180702、20180921;厂家B,批号:264180627、264180268、264180269;厂家C,批号:180801、180802、180803;规格均为0.4 g);奥拉西坦原料药[石药集团欧意药业(石家庄)有限公司,批号:264100627,纯度:99.3%];醋酸铵、冰醋酸、二氯甲烷、甲醇均为色谱纯,氢氧化钠、浓盐酸、磷酸二氢钠均为分析纯,水为超纯水。
2 方法与结果
2.1 奥拉西坦胶囊中主要杂质的定性分析
2.1.1 色谱条件 (1)一维液相色谱条件:色谱柱为ST PAK C18 ES柱(250 mm×4.6 mm,5 μm),流动相为0.02 mol/L磷酸二氢钠溶液,流速为0.5 mL/min,柱温为30 ℃,检测波长为210 nm,进样量为20 μL。(2)二维液相色谱条件和质谱条件:色谱柱为Techmate C18-STⅡ柱(150 mm×4.6 mm,5 μm),流动相为0.02 mol/L醋酸铵溶液(用冰醋酸调节pH为4.8),流速为0.5 mL/min,柱温为30 ℃;离子源为电喷雾离子源(ESI),离子源温度为500 ℃,接口电压为4 500 V(正离子模式)、-4 500 V(负离子模式),扫描方式为一级质谱全扫描(正、负离子模式),去簇电压为55 V(正离子模式)、-45 V(负离子模式)。
2.1.2 溶液的制备 (1)供试品溶液:精密称取奥拉西坦胶囊内容物适量,加0.02 mol/L磷酸二氢钠溶液溶解并稀释制成每1 mL中含奥拉西坦1 mg的溶液,即得。(2)自身对照溶液:精密量取上述供试品溶液0.5 mL,置于100 mL量瓶中,加0.02 mol/L磷酸二氢钠溶液稀释至刻度,摇匀,即得。
2.1.3 主要未知杂质定性分析 精密量取“2.1.2”项下自身对照溶液和供试品溶液各20 μL,按“2.1.1”项下条件注入液相色谱仪,经一维液相色谱条件分离确定主要未知杂质的保留时间为6.607 min,详见图2。将该未知杂质(保留时间为6.607 min的部分)切换到LOOP环中,进行二维脱盐分析,记录总离子流图和未知杂质质谱图,详见图3、图4。结果显示,在奥拉西坦一维液相色谱图中保留时间为6.607 min的未知杂质峰,其分子离子峰分别为m/z 159.9[M+H]+和m/z 158.1[M-H]-,分子量为159。采用2D-LC-IT-TOF/MS系统的LCMS Solution Ver 3.70 色谱工作站中的分子式预测模块“Accurate Mass Calculator”,结合化合物氢碳比、氮规则、质谱信息以及奥拉西坦杂质合成路线[9],预测该杂质的可能分子式为C6H9NO4,初步推断该杂质为奥拉西坦酸(化学结构式见图1)。
2.1.4 破坏试验 取奥拉西坦原料药适量,经0.2 mol/L盐酸溶液或0.2 mol/L氢氧化钠溶液分别进行酸、碱破坏后,按“2.1.2(1)”项下方法制成两种破坏样品供试品溶液后,再按“2.1.1(1)”项下色谱条件进样测定(图略)。结果显示,在一维色谱图保留时间约6.6 min处的色谱峰明显增大,经二极管阵列检测器检测后显示,其最大吸收波长为197 nm;然后按“2.1.1(2)”项下条件检测后显示,该杂质分子量为159,推测其为奥拉西坦在酸、碱作用下水解生成的奥拉西坦酸。
2.2 杂质奥拉西坦酸的制备纯化、标化及结构确证
2.2.1 杂质奥拉西坦酸的制备纯化 参考文献方法[9]进行制备与纯化。取奥拉西坦原料药20 g,置于500 mL回流瓶中,加入0.5 mol/L氢氧化钠溶液100 mL,搅拌使溶解,加热保温回流反应5 h后停止;冰浴降温至0 ℃后,用1 mol/L盐酸溶液调节pH为4,在50 ℃条件下蒸发至近干后,采用Sep-Pak?Silica固相萃取小柱层析,以二氯甲烷和甲醇梯度洗脱;收集洗脱液,在30 ℃条件下蒸发至干,得黄色油状物。取该油状物,加入90%甲醇50 mL后,在60 ℃条件下加热使溶解,趁热过滤;滤液于0 ℃条件下放置过夜,析出结晶后滤过;结晶以甲醇清洗后,在60 ℃条件下干燥,得白色固体,即得杂质奥拉西坦酸精制品。
2.2.2 奥拉西坦酸的标化及结构确证 取“2.2.1”项下制备纯化所得的奥拉西坦酸精制品适量,加0.02 mol/L磷酸二氢钠溶液溶解制成每1 mL中含奥拉西坦酸1 mg的溶液,再按“2.1.1(1)”项下色谱条件进样测定。采用峰面积归一化计算,结果所制奥拉西坦酸的含量为99.5%(n=6)。经1H-NMR(400 MHz,D2O)分析,结果显示,δ:4.40(m,1H),4.14(d,2H,J=17.6 Hz),3.92(dd,1H,J=17.6 Hz,6.5 Hz),3.42(dd,1H,J=10.4 Hz,1.9 Hz),2.87(dd,1H,J=17.2 Hz,6.4 Hz),2.28(dd,1H,J=17.2 Hz,2.0 Hz)。
2.3 奥拉西坦胶囊中奥拉西坦酸的含量测定
采用HPLC法进行含量测定。
2.3.1 色谱条件 采用“2.1.1(1)”项下色谱条件。
2.3.2 溶液的制备 (1)供试品溶液:按“2.1.2(1)”项下方法配制,即得。(2)自身对照溶液:取上述供试品溶液按“2.1.2(2)”项下方法配制,即得。(3)杂质对照品溶液:精密称取“2.2.1”项下奥拉西坦酸精制品25 mg,置于25 mL量瓶中,加0.02 mol/L磷酸二氢钠溶液溶解并稀释至刻度,摇匀,作为贮备液;临用前取该贮备液0.5 mL,置于100 mL量瓶中,加0.02 mol/L磷酸二氫钠溶液溶解并稀释至刻度,摇匀,即得。(4)空白辅料溶液;取胶囊辅料(本课题组根据各厂家处方自行配制)按照处方比例混合,精密称取适量,加0.02 mol/L磷酸二氢钠溶液溶解制成空白辅料溶液。
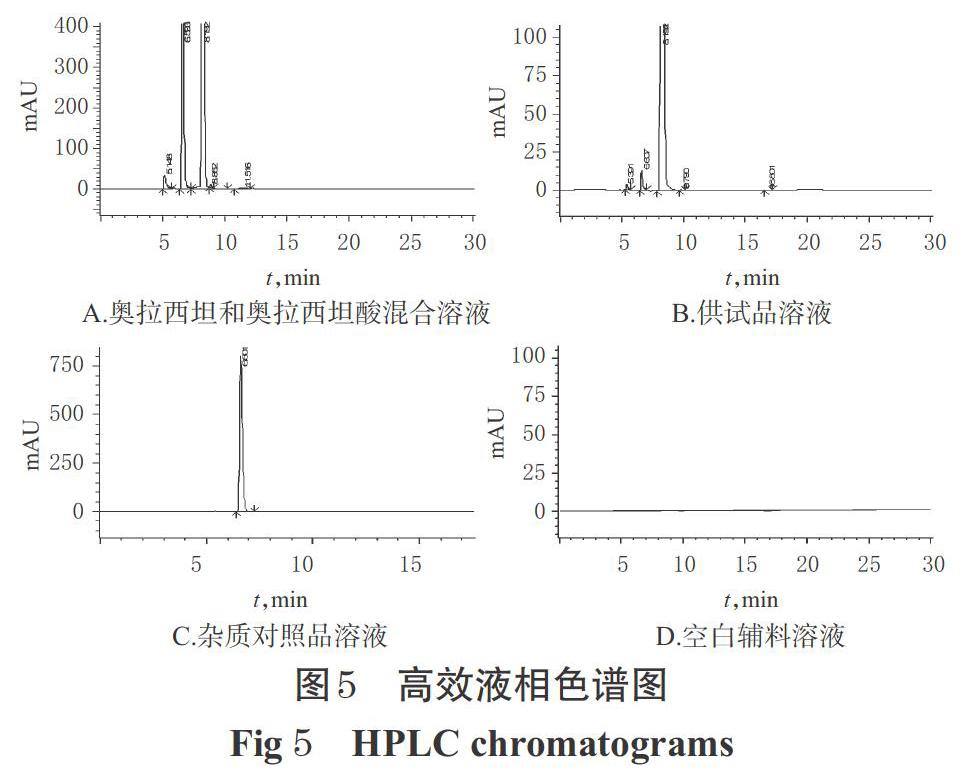
2.3.3 系统适用性试验 称取“2.2.1”项下奥拉西坦酸精制品适量,加入“2.3.2(2)”项下自身对照溶液适量,加0.02 mol/L磷酸二氢钠溶液稀释制成含奥拉西坦1 μg/mL、奥拉西坦酸5 μg/mL的混合溶液,再按“2.1.1(1)”项下色谱条件进样测定;另取“2.3.2(1)(3)(4)”项下供试品溶液、杂质对照品溶液、空白辅料溶液同法进样测定,记录色谱图,详见图5。结果显示,奥拉西坦和奥拉西坦酸分离度大于4.6,理论板数按奥拉西坦峰计不低于5 000,空白辅料无干扰。
2.3.4 检出限和定量限考察 取“2.3.2(3)”项下杂质对照品溶液,以0.02 mol/L磷酸二氢钠溶液为溶剂逐级稀释制得系列溶液,按“2.1.1(1)”项下色谱条件进样测定。结果,当信噪比分别为3 ∶ 1、10 ∶ 1时,得奥拉西坦酸的检出限为0.02 ng、定量限为0.1 ng。
2.3.5 线性关系考察 取“2.3.2(2)(3)”项下自身对照溶液和杂质对照品溶液,按“2.1.1(1)”项下色谱条件分别进样5、10、20、40、80 μL,记录峰面积。分别以奥拉西坦和奥拉西坦酸的进样量(X,ng)为横坐标、相应峰面积(Y)为纵坐标绘制标准曲线,得两者回归方程分别为:奥拉西坦Y=2 007.2X-2 647.2(r=1.000),奥拉西坦酸Y=2 821.0X-15 783.0(r=0.999 6)。结果表明,奥拉西坦和奥拉西坦酸进样量均在25~400 ng范围内与其峰面积呈良好的线性关系。
2.3.6 精密度试验 精密量取“2.3.3”项下奥拉西坦和奥拉西坦酸混合溶液 20 μL,按“2.1.1(1)”项下色谱条件连续进样测定6次,记录峰面积。结果,奥拉西坦和奥拉西坦酸峰面积的RSD分别为0.6%、0.9%(n=6),表明仪器精密度良好。
2.3.7 重复性试验 称取奥拉西坦胶囊内容物(批号:180802)适量,共6份,按“2.3.2(1)”项下方法制备供试品溶液,同时取“2.3.2(3)”项下杂质对照品溶液,分别按“2.1.1(1)”项下色谱条件进样测定,记录峰面积,按外标法计算奥拉西坦酸平均含量为0.05%(RSD=0.67%,n=6),表明本方法重复性良好。
2.3.8 加样回收率试验 称取奥拉西坦胶囊内容物(批号:180802)适量,共9份,精密称定,分别置于25 mL量瓶中,精密加入50 μg/mL杂质对照品溶液[按“2.3.2(3)”项下方法重新配制]0.8、1.0、1.2 mL,每个质量浓度平行3份。加样后,按“2.3.2(1)”项下方法制备供试品溶液,再按“2.1.1(1)”项下色谱条件进样测定,记录峰面积,计算加样回收率。结果,奥拉西坦酸平均加样回收率为99.25%(RSD=1.6%,n=9),详见表1。
2.3.9 稳定性试验 取同一份供试品溶液(批号:180802),分别于室温下放置0、2、4、6、8 h时,按“2.1.1(1)”项下色谱条件进样测定,记录峰面积。结果,奥拉西坦酸峰面积的RSD为0.96%(n=5),表明供试品溶液在室温下放置8 h内基本稳定。
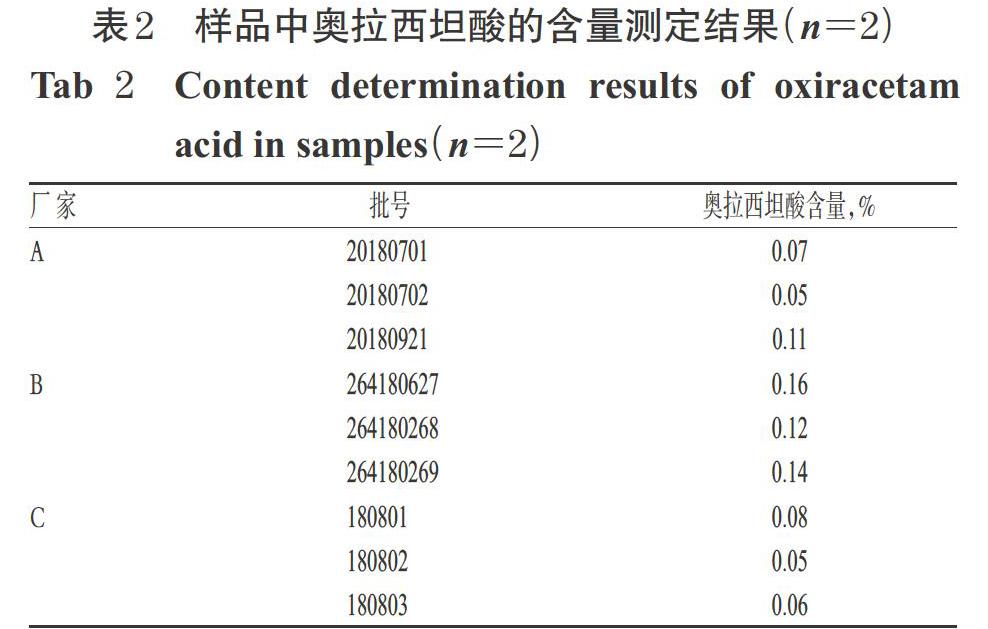
2.3.10 不同厂家9批样品中奥拉西坦酸的含量测定取3个厂家共9批奥拉西坦胶囊样品内容物适量,按“2.3.2(1)”项下方法制备供试品溶液,同时取“2.3.2(3)”项下杂质对照品溶液,分别按“2.1.1(1)”项下色谱条件进样测定,记录峰面积,采用外标法计算奥拉西坦酸含量(按胶囊规格0.4 g计算):杂质含量=(供试品溶液奥拉西坦酸峰面积×杂质对照品溶液浓度×样品平均装量)/(杂质对照品溶液峰面积×供试品溶液浓度×样品规格)×100%。每批样品平行2份测定,取平均值。结果,9批样品中均检出奥拉西坦酸,其测定结果详见表2。
3 讨论
3.1 色谱分析色谱柱的选择
现行奥拉西坦胶囊标准中无有关物质测定项,而其原料药注册质量标准中有关物质测定采用色谱柱为C8柱或氰基柱。本课题组在相同的色谱条件下,比较了C8柱、氰基柱和C18柱的保留时间和分离效果。结果显示,因奥拉西坦的极性大、分子小,故在C8柱上保留时间短、分离效果欠佳,主峰无法与破坏试验中产生的杂质峰较好地分离;采用氰基柱分离时,由酸、碱破坏试验所获得的杂质色谱峰较少,且主峰的纯度不高;采用ST PAK C18 ES色谱柱进行分析时,因该色谱柱进行了新型聚合物包被,不仅使奥拉西坦有适当的保留时间,而且对杂质的分离效果也好,故选择其作为一维色谱分析的色谱柱。
3.2 色谱分析流动相的选择
在确定了色谱柱的基础上,笔者考虑到需准确定位杂质的出峰时间,故重点对一维色谱流动相进行筛选。先后选用甲醇-水、乙腈-水、水、磷酸盐缓冲液等系统,并适当调节流动相比例后进行了一系列考察。结果显示,流动相中如果含有机溶剂,则柱效低、保留时间短,主峰与相邻峰不能得到较好分离;采用水为流动相则会出现较大的溶剂峰和倒峰;而采用磷酸盐缓冲液系统可避免出现以上情况。通过试验筛选后发现,以0.02 mol/L磷酸二氢钠缓冲盐为流动相时,主峰保留时间适宜,与各杂质峰的分离度良好,且峰宽较小,便于对目标杂质成分进行峰切割后转入二维色谱分析。
3.3 奥拉西坦杂质的产生
奥拉西坦含酰胺键,合成中在无水碳酸钠作用下易水解成羧酸,生成奥拉西坦酸(即4-羟基-2-氧代-1-吡咯烷乙酸)[10]。奥拉西坦酸为合成过程中的主要副产物,以及在酸性、碱性条件下的主要降解产物,可进一步与甲醇和乙醇发生酯化反应,生成4-羟基-2-氧代-1-吡咯烷乙酸甲酯和4-羟基-2-氧代-1-吡咯烷乙酸乙酯,所以控制奥拉西坦酸的含量显得尤为重要。
综上所述,奥拉西坦酸为奥拉西坦胶囊中的主要杂质;制备纯化后获得的奥拉西坦酸精制品含量达到99.5%,可以满足作为对照品的要求;所建立的超高效二维液相色谱-离子阱-飞行时间质谱法能准确定位杂质奥拉西坦酸的出峰位置并对其进行结构分析,且相应的含量测定方法能较好地分离杂质与主药及其他成分,灵敏度、精密度、重复性、稳定性、准确度均良好,符合2015年版《中国药典》要求[3]。因此,采用单一杂质外标法测定奥拉西坦酸的含量,并结合自身對照法测定其他杂质,可较好地跟踪测定生产制备过程中的中间体、控制奥拉西坦胶囊的成品质量。
参考文献
[ 1 ] 张倩倩.奥拉西坦治疗卒中后认知功能障碍的有效性及安全性探析[J].健康之路,2016,15(5):119-120.
[ 2 ] 金磊,李博,叶雷,等.奥拉西坦的临床前药理学研究[J].中国临床药理学与治疗学,2011,16(3):354-360.
[ 3 ] 国家药典委员会.中华人民共和国药典:四部[S]. 2015年版.北京:中国医药科技出版社,2015:354-356、374-378.
[ 4 ] 国家食品药品监督管理局.WS-1005(X-750)-2002 奥拉西坦:试行[S].2003-01-28.
[ 5 ] 国家食品药品监督管理局. YBH06612003 奥拉西坦:试行[S]. 2003-09-27.
[ 6 ] 国家食品药品监督管理局. YBH06622003 奥拉西坦胶囊:试行[S].2003-09-27.
[ 7 ] 沈保家,秦昆明,刘启迪,等.二维色谱技术及其在中药领域中的应用[J].中国科学(化学),2013,43(11):1480- 1489.
[ 8 ] 李郭帅,马阳,耿婷,等.超高效二维液相色谱法测定复方南星止痛膏中双酯型乌头生物碱的含量[J].药物分析杂志,2019,39(2):249-256.
[ 9 ] 张毅,陈瑞,汤磊.两个奥拉西坦杂质的合成[J].化学研究与应用,2018,30(4):602-604.
[10] 吴福鸿,李兆林,杨亚军.奥拉西坦的合成工艺改进[J].石油化工应用,2018,37(7):117-119.
(收稿日期:2019-08-01 修回日期:2019-02-12)
(编辑:段思怡)
猜你喜欢
中国药学药品知识仓库(2022年8期)2022-05-09
河南农业·综合版(2022年2期)2022-03-18
河南农业·综合版(2021年7期)2021-08-23
河南农业·综合版(2020年8期)2020-08-19
足球俱乐部(2018年19期)2018-10-30
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
中学生数理化·中考版(2015年12期)2015-09-10
科技致富向导(2013年15期)2013-09-09
